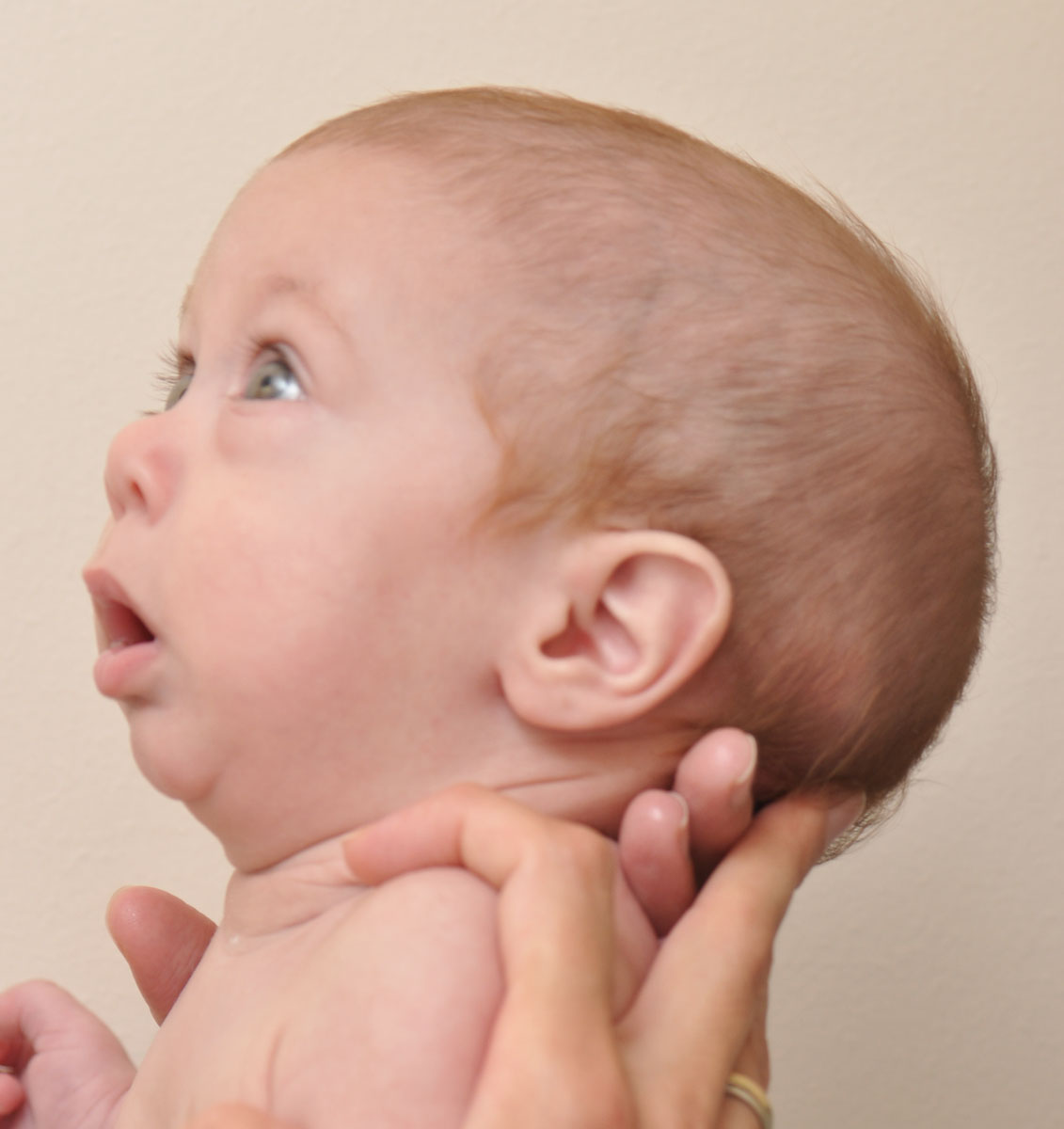
Что такое синдром Тричера-Коллинза?
Синдром Тричера-Коллинза (сокр. СТК, челюстно-лицевой дизостоз) — редкое генетическое заболевание, характеризующееся отличительными аномалиями головы и лица. Черепно-лицевые аномалии имеют тенденцию вызывать недоразвитие скулового комплекса, скул, челюстей, неба и рта, что может привести к затруднению дыхания и кормлению. Кроме того, у больных могут быть пороки развития глаз, включая наклоненные вниз отверстия между верхними и нижними веками (глазные щели) и аномалии структур наружного и среднего уха, которые могут привести к потере слуха.
Мозг и поведенческие аномалии, такие как микроцефалия и психомоторная задержка, также иногда наблюдаются при синдроме. Конкретные симптомы и физические признаки, связанные с СТК, могут сильно различаться. Некоторые люди могут быть настолько слабо затронуты болезнью, что их состояние может остаться не диагностированным, в то время как у других могут развиться серьезные, угрожающие жизни осложнения.
Хотя СТК в первую очередь возникает в результате изменения (мутации) в гене TCOF1, она также связана с мутациями в генах POLR1B, POLR1C или POLR1D. В случае TCOF1 и POLR1B тип наследования является аутосомно-доминантным (вид наследования, при котором генетическая обусловленная болезнь проявляется в случае, если у человека есть хотя бы один соответствующий ей «дефектный» ген, причем этот ген не содержится в половых (Х и Y) хромосомах), в то время как POLR1C наследуется по аутосомно-рецессивному типу (вид наследования, при котором генетически обусловленная болезнь проявляется в том и только в том случае, если «дефектный» ген был унаследован от обоих родителей и при этом не содержится в половых (Х и Y) хромосомах). Напротив, мутация в гене POLR1D, может быть как аутосомно-доминантная, так и аутосомно-рецессивная.
Синдром назван в честь Эдварда-Тричера-Коллинза, лондонского офтальмолога, который впервые описал это расстройство в медицинской литературе в 1900 году. СТК также известен как челюстно-лицевой дизостоз или синдром Тричера-Коллинза-Франческетти.
Признаки и симптомы
Симптомы и тяжесть синдрома Тричера-Коллинза могут значительно варьироваться от одного человека к другому, даже среди членов одной семьи. У некоторых людей симптомы могут быть слабовыражены, другие могут иметь значительные нарушения и потенциально опасные для жизни респираторные осложнения. Важно отметить, что у пострадавших людей не будет всех симптомов, обсуждаемых ниже.
Основные характерные черты СТК охватывают определенные кости лица, ушей и мягких тканей вокруг глаз. Пострадавшие люди имеют отличительные черты лица и потенциально могут иметь проблемы со слухом и зрением. Нарушения при СТК, как правило, симметричны (почти идентичны с обеих сторон лица) и присутствуют при рождении (врожденные). Развитие речи и языка может быть нарушено потерей слуха, расщелиной неба или челюсти и проблемами с дыхательными путями. На интеллект они обычно не влияют, но аномалии мозга и поведения, такие как микроцефалия и когнитивная задержка наблюдаются часто.
У детей с синдромом наблюдаются недоразвитость или отсутствие скул, в результате чего эта область лица выглядит плоской или впалой. Кость нижней челюсти развивается не полностью (гипоплазия нижней челюсти), в результате чего подбородок и нижняя челюсть выглядят аномально маленькими (микрогнатия). Некоторые костные структуры (например, венечные и кондилоидные отростки), которые соединяют участки кости нижней челюсти к мышце, могут быть необычно плоскими или отсутствовать. У пострадавших детей может также наблюдаться недоразвитость горла (гипоплазия глотки). Гипоплазия глотки с недоразвитостью нижней челюсти (гипоплазия нижней челюсти) и/или аномальная малость челюсти (микрогнатия) может способствовать проблемам с питанием и/или затруднению дыхания (дыхательная недостаточность) в раннем детстве. Дети могут испытывать обструктивное апноэ во сне, для которого характерны повторяющиеся кратковременные нарушения нормального дыхания и движения воздуха во время сна. У некоторых больных с серьезными поражениями могут возникнуть опасные для жизни проблемы с дыханием.
Дополнительные нарушения, которые могут способствовать затруднению дыхания или кормлению, включают сужение или обструкцию носовых дыхательных путей. Некоторые дети могут иметь признаки «последовательности Пьера Робина», которые включают тяжелую микрогнатию, язык, который смещен дальше назад во рту, чем обычно (глоссоптоз), с неполным закрытием крыши рта (волчья пасть) или без него. Даже у пациентов, где нёбо срастается, оно может оставаться выпуклым, что может повлиять на питание и дыхание. Кроме того, пороки развития рта и челюсти могут привести к зубным аномалиям, таким как неправильное прикус. Также сообщалось о дополнительных зубных аномалиях, включая отсутствие зубов.
У людей с СТК может развиться потеря слуха из-за неспособности звуковых волн проходить через среднее ухо (кондуктивная потеря слуха). Кондуктивная потеря слуха обычно возникает из-за аномалий, влияющих на структуры в среднем ухе, и у людей с СТК также могут быть неправильно сформированные или отсутствующие косточки, три маленьких кости, через которые звуковые волны передаются в среднее ухо. Кроме того, наружные ушные структуры часто отсутствуют. Внешние уши могут быть смяты или повернуты. Напротив, внутреннее ухо обычно не поражено, хотя сообщалось о мальформации костного спирального органа во внутреннем ухе (улитки) и структур во внутреннем ухе, которые играют роль в равновесии (вестибулярный аппарат). Дополнительные симптомы могут включать присутствие небольших наростов кожи или ямок непосредственно перед наружным ухом (доаурикулярные метки) и ненормального прохода, который закрыт на одном конце (слепой свищ), который обычно отводит уши к носу.
Многие дети с СТК имеют аномалии тканей, окружающих глаза. Эти различия в глазах могут дать пострадавшим пациентам печальный внешний вид лица. Наиболее распространенным глазным симптомом является наклон вниз к отверстию между верхним и нижним веками (глазные щели). Дополнительные симптомы включают в себя выемку нижнего века или расщелину отсутствующей ткани век (колобома век), частичное отсутствие ресниц на нижнем веке, косоглазие и суженные слезные протоки (дакростеноз). Иногда наблюдаются пороки развития глазного яблока, которые могут включать надрез или расщелину отсутствующей ткани радужной оболочки или аномально маленьких глаз (микрофтальмия). У некоторых пациентов может возникнуть потеря зрения. Степень нарушения зрения варьируется в зависимости от тяжести и сочетания глазных аномалий. Нарушение нижнего века может привести к высыханию глаз, что увеличивает риск хронического раздражения и глазных инфекций.
Приблизительно у 5% людей с СТК наблюдается дефицит развития или неврологические проблемы, такие как психомоторная задержка. Однако интеллект, как правило, не влияет на нормальное развитие языка. Тем не менее, проблемы с развитием речи могут возникать из-за потери слуха, расщепления нёба или трудностей с воспроизведением звуков из-за структурных искажений. У некоторых больных с СТК наблюдаются дополнительные физические отклонения, такие как широко расставленные глаза, насечка верхнего века, деформация носа, ненормально широкий рот (макростомия), необычный рост волос на голове в направлении щек, врожденные пороки сердца и/или желудочно-кишечные пороки развития.
Причины
СТК вызывается мутацией генов TCOF1, POLR1B, POLR1C или POLR1D. В случае мутации гена TCOF1 тип наследования является аутосомно-доминантным, хотя наблюдались очень редкие случаи аутосомно-рецессивных мутаций. Мутации POLR1B являются аутосомно-доминантными, тогда как при POLR1C они являются аутосомно-рецессивными, а мутации POLR1D могут быть аутосомно-доминантными или аутосомно-рецессивными.
Генетические заболевания определяются сочетанием генов для определенного признака, которые находятся на хромосомах, полученных от отца и матери. Доминантные генетические нарушения возникают, когда для появления заболевания необходима только одна копия ненормального гена. Для TCOF1, POLR1B и POLR1D аномальный ген может быть унаследован от любого из родителей или может быть результатом новой мутации (спонтанного изменения гена) у пострадавшего человека. Примерно у 60% пациентов с синдромом Тричера-Коллинза мутация является новой, которая возникает случайно (спонтанно) без предшествующего семейного анамнеза заболевания (мутация de novo). Тем не менее, родители также могут быть слегка затронуты и не знать, что у них есть расстройство. Риск передачи ненормального гена от пострадавшего родителя к потомству составляет 50% при каждой беременности. Риск одинаков для детей мужского и женского пола. Независимо от того, наследуется ли мутация от матери или отца, похоже, что она не имеет отношения к тяжести состояния СТК у их детей.
Рецессивные генетические нарушения (например, СТК, вызванные мутациями POLR1C или POLR1D) возникают, когда человек наследует один и тот же аномальный ген для одного и того же признака от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, человек будет носителем заболевания, но обычно не проявлять симптомов. Риск для двух родителей-носителей, чтобы передать оба дефектных гена и, следовательно, родить больного ребенка, составляет 25% при каждой беременности. Риск зачать ребенка, который будет носителем, как родители, составляет 50% при каждой беременности. Вероятность для ребенка получить нормальные гены от обоих родителей и быть генетически незатронутым для этой конкретной болезни составляет 25%.
Мутации гена TCOF1 вызывают большинство (приблизительно 80%) случаев синдрома Тричера-Коллинза. TCOF1 содержит инструкции, которые кодируют (создают) белок, известный как treacle. Точная роль, которую играет белок treacle в развитии СТК, полностью не понята. Исследователи определили, что treacle играет роль в создании определенных небольших структур в клетках, которые собирают белки (рибосомы). Это особенно важно для формирования группы клеток, называемых клетками нервного гребня, которые образуются очень рано во время эмбрионального развития и дают начало большей части кости и хряща, лежащих под лицом.
Состояния, которые возникают из-за дефектов в образовании (биогенезе) рибосом, называются рибосомопатиями. POLR1B кодирует субъединицу РНК-полимеразы 1, тогда как POLR1C и POLR1D кодируют субъединицы РНК-полимеразы I и III, каждая из которых также важна для биогенеза рибосом. Вероятно, что мутации в TCOF1, POLR1B, POLR1C и POLR1D вызывают недостаточную сборку белка и не позволяют определенным клеткам нервной системы и нервного гребня удовлетворять свои потребности в пролиферации и росте во время развития эмбриона. Поскольку СТК сильно различается, исследователи предполагают, что дополнительные генетические и, возможно, экологические факторы также могут играть роль в варьируемой степени тяжести заболевания. В поддержку этой концепции недавние экспериментальные данные показали, что белок treacle играет критическую роль в защите от вызванного окислительным стрессом повреждения ДНК в нервных клетках, а также в ориентации гребня во время деления нервных клеток, которые впоследствии влияют на развитие головы и лица.
Затронутые группы населения
Синдром Тричера-Коллинза затрагивает как мужчин, так и женщин в равных количествах. По оценкам, распространенность составляет от 1 на 10 000-50 000 человек в общей популяции. Некоторые люди с легкими проявлениями болезни могут не диагностироваться, что затрудняет определение истинной частоты расстройства в общей популяции. Поэтому настоятельно рекомендуется, чтобы родители ребенка и, возможно, братья и сестры страдающего от СТК в связи с мутацией в генах TCOF1, POLR1B, POLR1C или POLR1D, были проверенны, даже если кажутся здоровыми. Это важно для будущего планирования семьи. Не следует предполагать, что мутация у больного ребенка произошла самопроизвольно, просто потому, что у родителей нет лицевых различий. Тем не менее, следует отметить, что у некоторых людей (приблизительно 10-15%) с особенностями и физическими признаками СТК нет мутаций ни в одном из четырех вышеупомянутых генов, что позволяет предположить, что дополнительные, еще не идентифицированные гены также могут вызывать СТК.
Диагностика
Диагноз СТК ставится на основании тщательной клинической оценки, подробного анамнеза пациента и определения характерных физических признаков. При рождении присутствуют многие сопутствующие аномалии, такие как пороки развития или отсутствие наружного уха.
— Клиническое тестирование и обследование.
Специализированные рентгенологические исследования подтвердят наличие и/или степень определенных наблюдаемых черепно-лицевых аномалий. Например, такие визуальные исследования показывают аномальную малость челюсти (микрогнатия) из-за недоразвития кости нижней челюсти (гипоплазия нижней челюсти), наличие и/или степень гипоплазии, затрагивающей определенные части черепа, и/или наличие дополнительных пороков развития уха, которые нельзя увидеть во время клинической оценки.
Кроме того, у тех больных, у которых наблюдается мало признаков, тщательное клиническое обследование и рентгенография черепно-лицевой области могут продемонстрировать едва различимое присутствие определенных характерных признаков (например, гипоплазию скуловых дуг), связанных с синдромом. Поскольку синдром Тричера-Коллинза обладает несколькими физическими особенностями, которые могут встречаться при других черепно-лицевых синдромах, многие исследователи рекомендуют делать диагностическое подтверждение посредством молекулярно-генетического тестирования и/или, в некоторых случаях, тщательной, детальной семейной истории.
Молекулярно-генетическое тестирование для подтверждения диагноза доступно в коммерческих и академических исследовательских лабораториях для выявления мутаций в генах TCOF1, POLR1B, POLR1C и POLR1D. Приблизительно 80% людей имеют идентифицируемую мутацию гена TCOF1. Кроме того, генетическое подтверждение TCOF1, POLR1B, POLR1C или POLR1D мутаций может быть обнаружена до рождения (пренатально) путем амниоцентеза и взятия проб ворсинчатого хориона, если мутация была выявлена у больного члена семьи. В некоторых случаях ультразвуковое исследование плода, в котором используются отраженные звуковые волны для создания изображения развивающегося плода, может выявить характерные признаки, указывающие на наличие СТК у ребенка. Родственники, особенно родители и братья или сестры, лиц с диагнозом СТК должны быть тщательно обследованы, поскольку легкие случаи часто остаются нераспознанными и не диагностируемыми.
Стандартные методы лечения
От синдрома Тричера-Коллинза нет никакого лекарства. Лечение направлено на конкретные симптомы, которые проявляются у каждого человека. Лечение может потребовать скоординированных усилий группы специалистов. Педиатры, специалисты по лечению уха, носа и горла (детские отоларингологи), детские стоматологи, медсестры, пластические хирурги, логопеды, аудиологи, офтальмологи, психологи, генетики и другие медицинские работники может потребоваться систематическое и всестороннее планирование лечения ребенка, затронутого болезнью.
Врачи регулярно проводят мониторинг лиц с СТК для выявления определенных отклонений, которые могут быть связаны с расстройством. Например, следует внимательно следить за слухом больного, страдающего от СТК, чтобы выявить любые признаки потери слуха. Оценка слуха младенца имеет решающее значение, и полную оценку следует проводить в раннем возрасте, даже до одного года, а затем ежегодно, чтобы обеспечить правильное развитие речи.
Для визуализации внутренней части глаза используется инструмент (офтальмоскоп), позволяющий обнаружить любое возможное ухудшение зрения. Этот осмотр важен для обеспечения соответствующих профилактических мер и/или оперативного лечения больных, у которых наблюдаются отклонения в глазах в связи с синдромом Тричера-Коллинза (например, колобомы, косоглазия, микрофтальмии). Пострадавшие люди также должны находиться под наблюдением на предмет патологий челюсти и зубов.
Раннее вмешательство важно для обеспечения того, чтобы затронутые дети раскрыли свой потенциал. Специальные услуги, которые могут быть полезными, включают логопедическую, специальную социальную поддержку и другие медицинские, социальные и/или профессиональные услуги.
Прогноз
Обычно люди с синдромом Тричера-Коллинза становятся полноценными взрослыми с нормальным интеллектом. При правильном обращении и лечении ребенка ожидаемая продолжительность жизни примерно такая же, как и у населения в целом. В некоторых случаях прогноз зависит от конкретных симптомов и тяжести больного. Например, очень тяжелые случаи СТК могут вызвать перинатальную смерть из-за нарушения дыхательных путей.