Что такое синдром Барта?
Синдром Барта — это редкое метаболическое и нервно-мышечное генетическое заболевание, которое встречается исключительно у мужчин, поскольку оно передается от матери к сыну через Х-хромосому. Хотя синдром Барта обычно проявляется в младенчестве или в раннем детстве, возраст появления, связанные с ним симптомы и результаты, а также течение болезни значительно различаются даже среди затронутых членов одной семьи.
Основные характеристики расстройства включают аномалии сердечной и скелетной мышц (кардиоскелетная миопатия), низкий уровень определенных лейкоцитов (нейтрофилов, нейтропения), которые помогают бороться с бактериальными инфекциями, и замедление роста, потенциально приводящее к низкому росту. Нарушение также связано с повышенным уровнем некоторых органических кислот в моче и крови, таких как 3-метилглутоновая ацидурия/ацидемия. Левый желудочек сердца может иметь увеличенную толщину в результате необычно высоких концентраций эластичных коллагеновых волокон (эндокардиальный фиброэластоз). Утолщение уменьшает способность левого желудочка выталкивать кровь через легкие и, таким образом, является основным источником потенциальной сердечной недостаточности.
Синдром Барта передается в виде Х-сцепленной рецессивной черты. Ген, ответственный за расстройство, был расположен на длинном плече (q) хромосомы X в Xq28.
Признаки и симптомы
Симптомы, связанные с синдромом Барта, могут быть очевидны при рождении, младенчестве или раннем детстве. Тем не менее, в редких случаях расстройство не может быть диагностировано до зрелого возраста.
У большинства людей с синдромом Барта наблюдается ослабление сердечной мышцы (кардиомиопатия), что приводит к расширению нижних отделов сердца (желудочков).
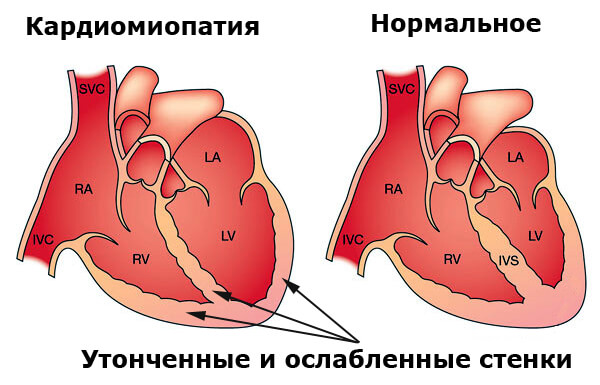
Известные как дилатационная кардиомиопатия, признаки этого состояния часто присутствуют при рождении или могут появляться в течение первых месяцев жизни. Дилатационная эндокардиальная миопатия, как правило, ослабляет насосную деятельность сердца, уменьшая объем крови, циркулирующей в легких и остальной части тела, вызывая сердечную недостаточность. Симптомы сердечной недостаточности могут зависеть от возраста ребенка и других факторов. Например, у маленьких детей сердечная недостаточность может проявляться в виде усталости и одышки при физической нагрузке.
Синдром Барта также связан с ненормально сниженным мышечным тонусом (гипотонией) и мышечной слабостью (скелетной миопатией), что часто приводит к задержкам в приобретении грубых двигательных навыков. Общие моторные навыки включают такие действия, как ползание, ходьба, бег, прыжки и поддержание равновесия. Это навыки, которые требуют использования и координации больших групп мышц. Слабость мимических мышц может привести к необычным выражениям лица.
Кроме того, пострадавшие младенцы и дети могут не развиваться и не набирать вес с ожидаемой скоростью. У них могут быть слабые нарушения обучения (хотя они обычно имеют нормальный интеллект), и во многих случаях могут быть подвержены рецидивирующим бактериальным инфекциям из-за низкого уровня циркулирующих нейтрофилов в крови.
Без своевременного выявления и соответствующего лечения сердечная недостаточность и бактериальные инфекции могут быть опасными для жизни осложнениями.
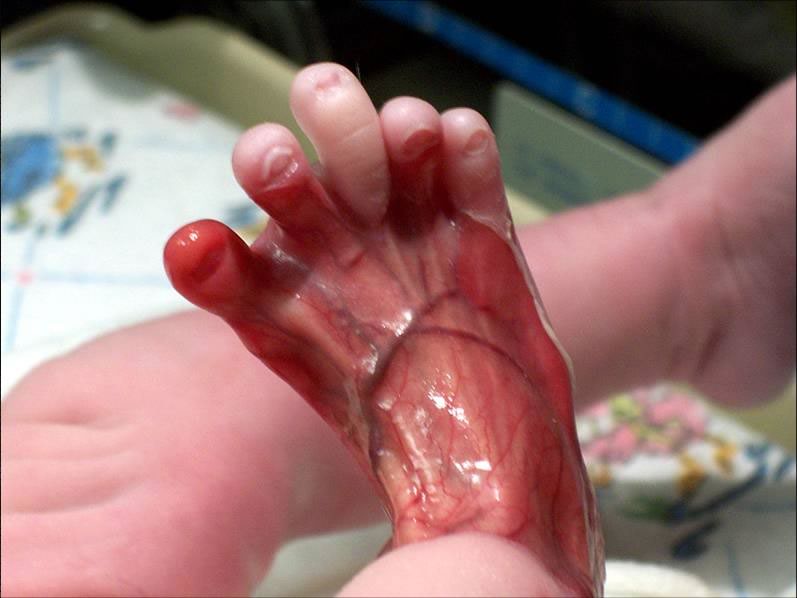
На фото выше врожденная аплазия кожи при синдроме Барта.
В дополнение к аномалиям сердечной и скелетной мышц, нейтропении и задержке роста, у пациентов с синдромом Барта есть специфический биохимический маркер, который в течение многих лет распознавался как основной индикатор синдрома Барта.
Биохимический маркер — это любое вещество, такое как фермент или малая молекула, которое обнаруживается в моче или других жидкостях организма и служит диагностическим признаком конкретного расстройства.
Исследователи показали, что люди с синдромом Барта имеют аномально повышенные уровни 3-метилглутаконовой кислоты в моче и в жидкой части крови. По словам клиницистов, дети с синдромом Барта могут иметь повышенный уровень 3-метилглутаконовой кислоты в крови с середины младенчества до примерно трехлетнего возраста. Однако связи между повышенным содержанием кислоты и тяжестью других симптомов и признаков, связанных с синдромом Барта, не прослеживается.
Под микроскопом клетки сердечной мышцы пациентов с синдромом Барта имеют митохондрии аномальной формы. Другими метаболическими признаками, которые сами по себе не являются диагностическими, но служат для подтверждения диагноза, основанного на других критериях, являются высокий уровень молочной кислоты в крови и моче (побочный продукт интенсивной мышечной активности) и низкий уровень карнитина. Карнитин играет роль в движении химических веществ, особенно жирных кислот, через клеточную мембрану.
Причины синдром Барта

Синдром Барта передается как рецессивный признак, связанный с Х-образной связью. Неисправный ген прослеживается до участка хромосомы X на карте гена локуса Xq28.
Хромосомы, которые присутствуют в ядре клеток человека, несут генетическую информацию каждого человека. Клетки человеческого тела обычно имеют 46 хромосом. Пары человеческих хромосом пронумерованы от 1 до 22, а половые хромосомы обозначены X и Y. У мужчин есть одна X и одна Y-хромосома, а у женщин две X-хромосомы. Каждая хромосома имеет короткое плечо, обозначенное «p», и длинное плечо, обозначенное «q». Хромосомы далее подразделяются на множество полос, которые пронумерованы. Например, «хромосома Xq28» относится к полосе 28 на длинном плече Х-хромосомы. Пронумерованные полосы указывают местоположение тысяч генов, присутствующих в каждой хромосоме.
Генетические заболевания определяются сочетанием генов определенного признака, которые находятся на хромосомах, полученных от отца и матери.
Х-сцепленные рецессивные генетические нарушения представляют собой состояния, вызванные ненормальным геном в Х-хромосоме. У женщин две Х-хромосомы, но одна из Х-хромосом «выключена», и все гены в этой хромосоме инактивированы. Женщины, у которых есть ген заболевания, присутствующий в одной из их Х-хромосом, являются носителями этого расстройства. Женщины-носители обычно не проявляют симптомов расстройства, потому что это обычно Х-хромосома с ненормальным геном «выключен». У мужчины есть одна Х-хромосома, и если он унаследует Х-хромосому, содержащую ген болезни, у него разовьется болезнь. Мужчины с Х-связанными нарушениями передают ген болезни всем своим дочерям, которые будут носителями. Мужчина не может передать X-связанный ген своим сыновьям, потому что мужчины всегда передают свою Y-хромосому вместо своей X-хромосомы потомству мужского пола.
В некоторых случаях мать больного мужчины может не быть носителем синдрома Барта, и иметь очевидной семейной истории болезни. В таких случаях расстройство, по-видимому, является результатом новой мутации гена в Х-хромосоме, которая произошла случайно по неизвестным причинам (спорадически).
Ген, расположенный в хромосоме X28, известен как ген TAZ. Ген TAZ кодирует группу белков, называемых таффазинами, которые выполняют как минимум две функции. Во-первых, эти белки играют роль в поддержании внутренних, сильно свернутых мембран митохондрий в клетках. Митохондрии являются производящими энергию крупинками, от которых зависит клетка. Таффазины служат для обеспечения того, чтобы концентрация специфического жира (кардиолипина) была достаточной для поддержания выработки энергии в митохондриях. Таффазины также способствуют развитию костных клеток из клеток-предшественников кости (остеобласты).
Мутации в гене TAZ также ответственны за появление 3-метилглутаконовой кислоты в крови и моче пациентов с синдромом Барта.
Затронутые группы населения
Синдром Барта, кажется, затрагивает все этнические группы. Эпидемиологи из медицинского центра университета Джона Хопкинса оценивают частоту возникновения этого синдрома где-то между 1 на 200 000 — 400 000 новорожденных. Однако они предупреждают, что эти цифры могут быть недооценены, поскольку считают, что многие случаи синдрома Барта остаются неопознанными и не регистрируются.
Диагностика
Синдром Барта может быть диагностирован в младенчестве или в раннем детстве (или, в некоторых случаях, в более позднем возрасте), основываясь на тщательной клинической оценке, выявлении характерных физических данных, полной истории болезни пациента и семьи и разнообразных специализированных тестах.
Эксперты указывают, что диагноз синдрома Барта следует рассматривать для любого мужчины или ребенка с:
- дилатационной кардиомиопатией неизвестной причины (идиопатической);
- низким уровнем циркулирующих нейтрофилов (нейтропения);
- повышенным уровнем в моче 3-метилглутаконовой кислоты (ацидурия);
- аномальными митохондриями в сердечной мышце;
- и/или мышечными аномалиями (миопатией) неизвестной причины, возникающие в связи с задержкой роста.
Для младенцев и детей с признаками кардиомиопатии следует проводить метаболические скрининговые тесты, в том числе исследования измерения уровня 3-метилглутаконовой кислоты и других органических кислот в моче и крови.
Как упоминалось выше, повышенный уровень в моче 3-метилглутаконовой кислоты (3-метилглутаконовой ацидурии) был признан биохимическим маркером, который может функционировать как диагностический признак синдрома Барта. Важно также измерить концентрацию нейтрофилов в крови. Постоянный низкий уровень нейтрофилов помогает подтвердить диагноз в сочетании с этими другими признаками.
Стандартные методы лечения
Лечение синдрома Барта направлено на конкретные симптомы, которые проявляются у каждого человека. Такое поддерживающее лечение может потребовать скоординированных усилий команды медицинских специалистов, таких как педиатры; врачи, специализирующиеся на заболеваниях сердца у детей (детские кардиологи); специалисты по исследованию крови и кроветворных тканей (гематологи); специалисты по лечению бактериальных инфекций, физиотерапевты; трудотерапевты; и/или другие специалисты здравоохранения.
Сердечная недостаточность и/или бактериальные инфекции являются более серьезной угрозой для пациента с синдромом Барта. Многие младенцы и дети с синдромом Барта нуждаются в терапии диуретиками и дигиталисом для лечения сердечной недостаточности. Имеющиеся данные свидетельствуют о том, что многие больные дети могут постепенно исключаться из такой сердечной терапии в более позднем детстве из-за улучшения работы сердца.
Для пациентов с подтвержденной нейтропенией осложнения, вызванные бактериальной инфекцией, часто можно предотвратить путем постоянного мониторинга и ранней терапии подозреваемых инфекций антибиотиками. Например, антибиотики могут быть предоставлены в качестве профилактической терапии во время нейтропении, чтобы предотвратить возникновение инфекции. Для некоторых людей с нейтропенией, таких как люди с повторяющимися бактериальными инфекциями и уровнями нейтрофилов, которые постоянно ниже 500, врачи могут рекомендовать использование агентов, которые стимулируют выработку костного мозга лейкоцитами.
Генетическое консультирование также пойдет на пользу пострадавшим людям и их семьям. Другое лечение этого расстройства является симптоматическим и поддерживающим.
Прогноз
Ранний и точный диагноз является ключом к длительной выживаемости мальчиков, рожденных с синдромом Барта. Тяжелые инфекции и сердечная недостаточность являются частыми причинами смерти у пораженных детей.