Что такое синдром Секкеля?
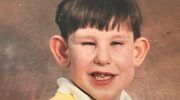
Синдром Секкеля чрезвычайно редкое наследственное заболевание, характеризующееся задержкой роста до рождения (задержка внутриутробного развития), что приводит к низкой массе тела при рождении. Задержка роста продолжается после рождения (послеродовой период), что приводит к низкорослости (карликовость). Другие симптомы и физические особенности, связанные с синдромом Секкеля, включают аномально маленькую голову (микроцефалия); разную степень умственной отсталости; и/или необычные характерные черты лица, включая «клювоподобный» выступ носа. Другие черты лица могут включать аномально большие глаза, узкое лицо, деформированные уши и/или необычно маленькую челюсть (микрогнатия). Кроме того, у некоторых младенцев может наблюдаться постоянная фиксация пятых пальцев в согнутом положении (клинодактилия), деформация (дисплазия) тазобедренного сустава, вывих кости в предплечье (радиальный смещение) и/или другие физические отклонения.
Признаки и симптомы
Синдром Секкеля характеризуется аномально медленным развитием плода и низкой массой тела при рождении. После рождения у ребенка будет наблюдаться медленный рост и созревание костей, что приведет к низкому, но пропорциональному росту (в отличие от карликовости коротких конечностей или ахондроплазии).
Больные с синдромом Секкеля имеют различные физические отклонения и особенности развития, в том числе:
- Очень маленький размер и вес при рождении (в среднем 3,3 фунта или 1,4 кг);
- Чрезвычайно маленький, пропорциональный рост;
- Аномально маленький размер головы (микроцефалия);
- Клювовидный выступ носа;
- Узкое лицо;
- Деформированные уши;
- Необычно маленькая челюсть (микрогнатия);
- Умственная отсталость, часто тяжелая, с IQ (коэффициент интеллекта) менее 50.
Другие симптомы могут включать аномально большие глаза, высокое арочное небо, пороки развития зубов и другие деформации костей. Также часто наблюдаются такие заболевания крови, как анемия (низкий уровень эритроцитов), панцитопения (недостаток клеток крови) или острый миелоидный лейкоз (тип рака крови).
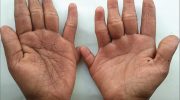
В некоторых случаях у мужчин яички не могут опускаться в мошонку (крипторхизм), а у женщин может быть ненормально увеличенный клитор. Кроме того, у больных с синдромом может быть чрезмерное оволосение на теле и одна единственная глубокая складка на ладонях (известная как обезьянья складка).
Причины
Синдром Секкеля — чрезвычайно редкое заболевание, передающееся по аутосомно-рецессивному типу. Три варианта синдрома Секкеля включают нарушения или изменения (мутации) генов на трех разных хромосомах. Расположение генной карты: синдром Секеля 1 на хромосоме 3 (3q22-q24); Синдром Секкеля 2 на хромосоме 18 (18p11.31-q11) и синдром Секеля 3 на хромосоме 14 (14q21-q22).
Хромосомы, присутствующие в ядре клеток человека, несут генетическую информацию для каждого человека. Клетки человеческого тела обычно имеют 46 хромосом. Пары человеческих хромосом пронумерованы от 1 до 22, а половые хромосомы обозначены X и Y. У мужчин одна X и одна Y хромосома, а у женщин две X хромосомы. У каждой хромосомы есть короткое плечо, обозначенное буквой «p», и длинное плечо, обозначенное буквой «q». Хромосомы далее подразделяются на множество пронумерованных полос. Например, «хромосома 3q22-q24» относится к области между полосой 22 и полосой 24 на длинном плече хромосомы 3. Подобным образом «хромосома 18p11.31-q11.2» относится к более широкой области между полосой 11p31 на коротком плече хромосомы 18 и полосе 11.2 на длинном плече хромосомы 18. Опять же, хромосома 14q21-q22 относится к более узкой области на длинном отрезке хромосомы 14 между полосами 21 и 22. Пронумерованные полосы указывают расположение тысяч генов, присутствующих на каждой хромосоме.
Специфический ген, участвующий в синдроме Секкеля 1, известен как ген атаксии-телеангиэктазии и ген Rad3-родственного белка (ATR). Гены, участвующие в синдроме Секкеля 2 и 3 типов, неизвестны.
Генетические заболевания определяются комбинацией генов определенного признака, которые находятся на хромосомах, полученных от отца и матери.
Рецессивные генетические расстройства возникают, когда человек наследует один и тот же аномальный ген одного и того же признака от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, он будет носителем болезни, но обычно бессимптомным. Риск для двух родителей-носителей передать дефектный ген и, следовательно, иметь больного ребенка, составляет 25% при каждой беременности. Риск иметь ребенка, который будет носителем, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей и будет генетически нормальным по данному признаку, составляет 25%. Риск одинаков для мужчин и женщин.
Все люди несут несколько аномальных генов. Родители, которые являются близкими родственниками (кровными родственниками), имеют больше шансов, чем неродственные родители, иметь один и тот же аномальный ген, что увеличивает риск рождения детей с рецессивным генетическим заболеванием.
Затронутые группы населения
Синдром Секкеля — чрезвычайно редкое наследственное заболевание, которое поражает в равной степени мужчин и женщин. Точная частота расстройства неизвестна. С момента первоначального описания в 1960 г. в медицинской литературе было зарегистрировано более 100 случаев.
Близкие по симптомам расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Секкеля. Сравнения могут быть полезны для дифференциальной диагностики:
Синдром Секкеля — один из шести расстройств в классе карликовости, известном как «изначальная карликовость». Эти расстройства имеют схожие характеристики, включая пороки развития скелета (дисплазию), дефицит роста до рождения (задержка внутриутробного развития) и в младенчестве и детстве, что в конечном итоге приводит к разной степени низкого роста. В эту группу заболеваний в настоящее время входят пять расстройств: синдром Секкеля, синдром Мейера-Горлина; Синдром Рассела-Сильвера; Остеодиспластическая примордиальная карликовость Маевского I/III; и остеодиспластическая примордиальная карликовость Маевского II типа.
Остеодиспластическая примордиальная карликовость Маевского II типа является чрезвычайно редким генетическим заболеванием, характеризующимся низким ростом, низкой массой тела при рождении, аномально маленькой головой (микроцефалия) и/или аномалиями скелета. Другие физические признаки могут включать большие глаза, выступающий нос в форме клюва, опущенную челюсть и/или узкое лицо. Обычно возникает серьезная недостаточность роста до рождения (внутриутробная недостаточность роста). В некоторых случаях может присутствовать умственная отсталость. Также могут появиться различные дополнительные симптомы. Конкретные симптомы и степень тяжести варьируются от случая к случаю. Считается, что II тип остеодиспластической примордиальной карликовости Маевского наследуется по аутосомно-рецессивному типу.
Диагностика
С появлением технически совершенной ультасонографии синдром Секкеля можно диагностировать до рождения (пренатально). При ультразвуковом исследовании плода используются отраженные звуковых волны для создания изображения развивающегося плода. После рождения можно заподозрить синдром Секкеля на основании тщательного клинического обследования, подробного анамнеза пациента и различных специализированных тестов. Хотя характерные черепно-лицевые, скелетные и/или другие физические аномалии, связанные с синдромом Секкеля, могут присутствовать при рождении, в некоторых случаях диагноз может не подтверждаться до тех пор, пока больной ребенок не вырастет и не разовьется полный синдром (например, когда станет очевидным низкий рост и/или умственная отсталость).
Низкий рост, связанный с синдромом Секкеля, предполагает пропорциональный рост рук и ног, что позволяет проводить дифференциальную диагностику от синдромов, связанных с низким ростом и аномально маленькими руками и ногами (карликовость с короткими конечностями).
Стандартные методы лечения
От синдрома Секкеля нет лекарства. Некоторые лекарства могут быть назначены для устранения других симптомов, связанных с заболеванием.
Лечение направлено на решение любой медицинской проблемы, которая может возникнуть, особенно заболеваний крови и структурных деформаций. Людям с умственными недостатками и их семьям потребуется соответствующая социальная поддержка и консультационные услуги.
Прогноз
Дети, страдающие синдромом Секкеля, могут жить в течение длительного периода времени, хотя они часто сталкиваются с глубокими психическими и физическими отклонениями.