Что такое болезнь Фабри?
Болезнь Фабри является редким наследственным нарушением метаболизма гликосфинголипидов (жиров), возникающим в результате отсутствия или заметно недостаточной активности лизосомального фермента, α-галактозидазы A (α-Gal A). Это расстройство относится к группе заболеваний, известных как лизосомные болезни накопления. Этот ферментативный дефицит вызван изменениями (мутациями) в α-галактозидазе A (GLA) гене, который инструктирует клетки производить фермент α-галактозидазу A (α-Gal A).
Лизосомы функционируют как первичные клетки пищеварительного тракта. Ферменты в лизосомах разрушают или переваривают определенные соединения и внутриклеточные структуры. α-Gal А функционирует, расщепляя сложные молекулы сахара-липида, называемых гликолипидами, в частности, глоботриаозилцерамидом (GL-3 или Gb3), его деацилированной формой лизо-GL-3/Gb3 и родственными гликолипидами, путем удаления концевого галактозного сахара с конца из этих гликолипидных молекул. Дефицит фермента вызывает непрерывное накопление GL-3/Gb3 и связанных с ним гликолипидов в клетках организма, что приводит к аномалиям клеток и дисфункции органов, которые особенно поражают мелкие кровеносные сосуды, сердце и почки.
Ген GLA расположен на Х-хромосоме, и поэтому болезнь Фабри наследуется как Х-сцепленное расстройство. Мужчины с классическим фенотипом типа 1 и фенотипами с поздним началом 2 типа, как правило, подвержены значительно более серьезным заболеваниям, чем женщины. У женщин, как правило, более изменчивое течение, и у них болезнь может протекать бессимптомно или иметь такие же серьезные последствия, как и у мужчин.
Существует два основных фенотипа заболевания: тип 1 «классический» и тип 2 «поздний». Оба приводят к почечной недостаточности и/или болезням сердца, а также к преждевременной смерти. Мужчины с 1 типом имеют небольшую или нулевую функциональную ферментативную активность α-Gal А (<3% от нормальной средней активности) и заметное накопление GL-3/Gb3 и родственных гликолипидов в капиллярах и мелких кровеносных сосудах, которые вызывают основные симптомы в детском возрасте или подростковый возраст. К ним относятся:
- акропарестезии (мучительная боль в руках и ногах, возникающая при физической нагрузке, лихорадке, стрессе и т.д.);
- ангиокератомы (кожные пятна от красного до синего цвета);
- ангидроз или гипогидроз (отсутствует или заметно снижается потоотделение);
- желудочно-кишечные симптомы, включая боль в животе и спазмы;
- дистрофия роговицы.
С увеличением возраста системное отложение GL-3/Gb3, особенно в сердце, приводит к аритмиям, гипертрофии левого желудочка, а затем к гипертрофической кардиомиопатии, а в почках к прогрессирующей протеинурии, почечной недостаточности и/или к цереброваскулярным заболеваниям, включая преходящие ишемические приступы (ТИА) и инсульты. До заместительной почечной терапии (т.е. диализа и трансплантации) и фермент-заместительной терапии средний возраст смерти у больных мужчин с классическим фенотипом 1 типа составлял ~ 40 лет. Заболеваемость мужчин с классическим фенотипом типа 1 составляет около 1 на 40000, но варьируется в зависимости от географического региона и расы, от ~ 1 на 18 000 до 1 на 95 000 на основе скрининговых исследований новорожденных.
Напротив, мужчины с фенотипом «позднего начала» типа 2 (ранее называвшиеся сердечными или почечными вариантами) имеют остаточную активность α-Gal А, испытывают недостаток накопления GL-3/Gb3 в капиллярах и мелких кровеносных сосудах и не показывают раннего проявления как при типе 1 (т.е. акропарестезии, гипогидроз, ангиокератомы, дистрофия роговицы и т.д.). Они в основном нормально чувствуют себя в детстве и юности и обычно страдают заболеваниями почек и/или сердца в течение третьего-седьмого десятилетий жизни.
Клинические проявления у гетерозиготных женщин из семей с классическим фенотипом типа 1 являются переменными вследствие случайной инактивации Х-хромосомы и варьируются от бессимптомных до таких же тяжелых, как у мужчин с классическим 1 типом, Гетерозиготы типа 2 могут быть бессимптомными или развить почечные или сердечные проявления в более позднем возрасте. Приблизительно 90% гетерозигот типа 1 имеют характерную дистрофию роговицы, в то время как у гетерозиготных женщин типа 2 обычно отсутствуют характерные признаки со стороны роговицы или другие ранние проявления типа 1. Частота и тяжесть проявлений у гетерозиготных женщин с 2 типом заболевания исследована систематически лишь в последнее время, и у них, как правило, симптомы встречаются реже и они менее тяжелые, чем у мужчин с 2 типом.
Признаки и симптомы
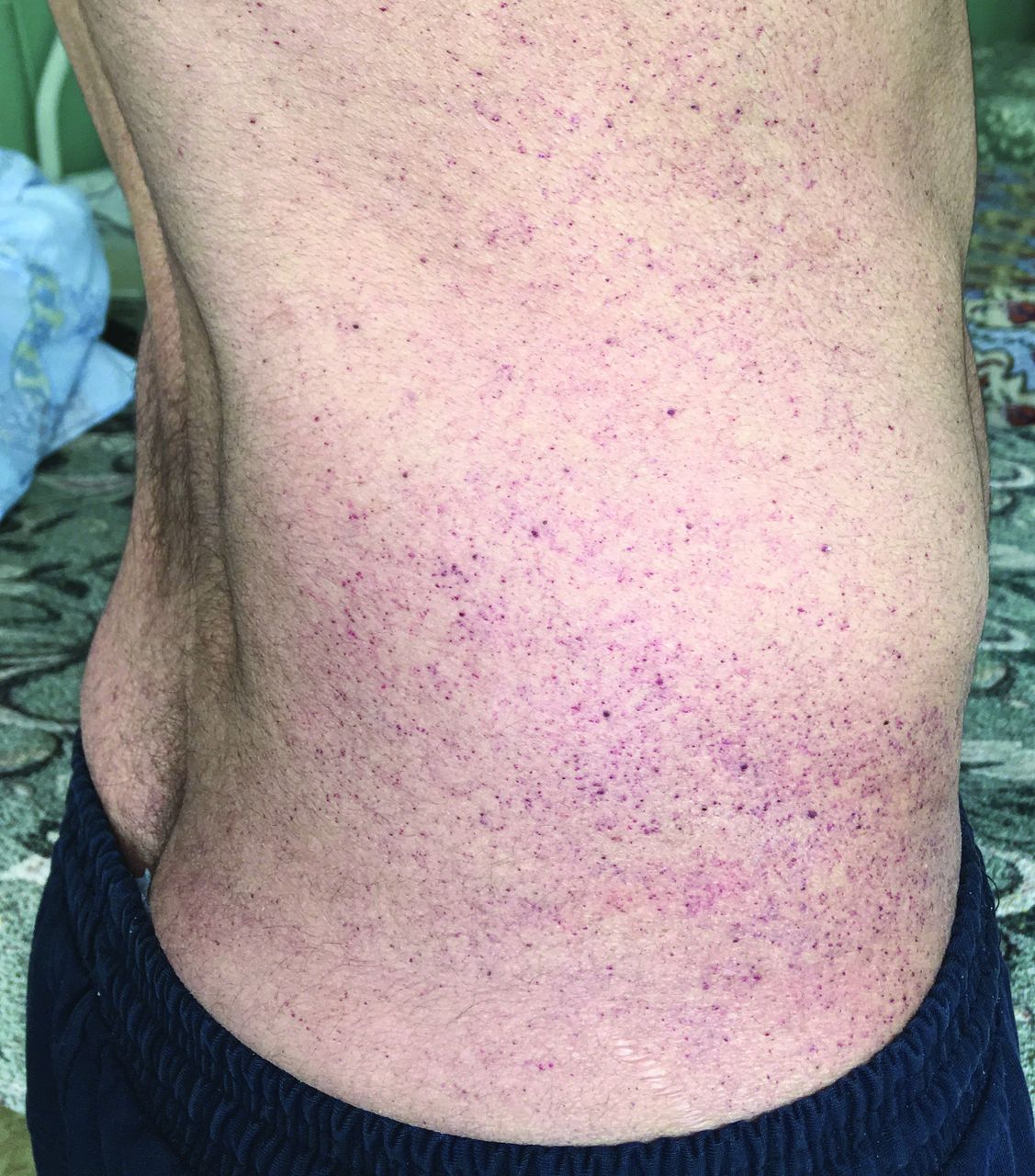
— Классический фенотип (тип 1).
Признаки и симптомы у мужчин с классическим фенотипом типа 1 обычно начинаются в детстве или в подростковом возрасте. Симптомы усиливаются с возрастом, главным образом, из-за прогрессирующего накопления гликолипидов в микрососудистой системе, подоцитах почек и кардиомиоцитах, приводящих к почечной недостаточности, болезням сердца и/или инсультам. Ранние и прогрессирующие клинические симптомы включают:
- Акропарестезии. Боль является ранним симптомом классического подтипа типа 1 и может встречаться у мужчин в возрасте 2-8 лет, а также может возникать в детском или подростковом возрасте у женщин-гетерозигот, особенно с лихорадкой. Пострадавшие могут испытывать эпизоды сильной жгучей боли в руках и ногах. Тяжелые приступы боли (кризисы Фабри) могут длиться от нескольких часов до нескольких дней и часто вызываются физическими нагрузками, усталостью, стрессом и/или лихорадкой.
- Ангидроз или гипогидроз. Мужчины и некоторые женщины с 1 типом имеют пониженную или отсутствующую выработку пота (гипогидроз или ангидроз) и дискомфорт при теплой температуре, при физической нагрузке или при лихорадке.
- Ангиокератома. Ранние симптомы также включают появление кожных высыпаний от красноватого до темно-синего цвета, особенно в области между бедрами и коленями (см. фото выше). Эти повреждения кожи могут быть плоскими или выпуклыми. Они часто встречаются в области пупка или половых органов мужчин с 1 типом заболевания. Как правило, у мужчин и женщин с фенотипом позднего типа 2 нет этих характерных поражений кожи.
- Желудочно-кишечные проблемы. Симптомы ЖКТ являются ранним проявлением заболевания Фабри 1-го типа. Могут также возникать спазмы в животе, частые движения кишечника и диарея, особенно после обильного приема пищи.
- Дистрофия роговицы. Пациенты с классическим фенотипом 1 типа имеют аномальные отложения гликолипидов в их роговице, что приводит к характерной мутности, что можно увидеть при осмотре с помощью щелевой лампы опытным офтальмологом. Эти изменения не влияют на зрение. Кровеносные сосуды в глазах могут казаться скрученными (в виде пробкового винта; искривленными) и/или слегка увеличенными (расширенными) из-за накопления гликолипидов в стенках сосудов.
Другие дополнительные симптомы, которые могут быть связаны с болезнью Фабри 1-го типа, включают:
- хроническую усталость;
- головокружение;
- головную боль;
- общую слабость;
- тошноту и/или рвоту;
- замедленное половое созревание;
- отсутствие или редкий рост волос;
- пороки развития суставов пальцев (редко).
У некоторых мужчин c классическим 1 типом наблюдается аномальное накопление лимфы в ступнях и голенях, связанное с отеком (лимфедема). У этих пациентов лимфа, жидкость организма, содержащая определенные белые кровяные клетки, жиры и белки, накапливается вне кровеносных сосудов в пространствах между клетками и дренажами или попадает обратно в кровоток через лимфатические сосуды. Лимфедема возникает в результате нарушения нормального дренажа (оттока) лимфы из-за накопления гликолипидов в лимфатических сосудах и лимфатических узлах.
— Распространенные проявления у мужчин с 1 и 2 типом заболевания.
С возрастом у мужчин с 1 типом расстройства, обычно в третьем-четвертом десятилетиях, и у мужчин с 2 типом в третьем-шестом десятилетиях прогрессирующее отложение гликолипидов GL-3/Gb3 приводит к почечным и/или сердечным проявлениям, как описано ниже. Многие из мужчин с поздним началом 2 типа, у которых отсутствуют ранние проявления, наблюдаемые при 1 типе заболевания, обнаруживают проблемы почек, сердца или инсульты.
Пациенты с более поздним подтипом типа 2 обычно не имеют кожных поражений (ангиокератома), нормально потеют, не испытывают болей Фабри или кризов и не имеют непереносимости тепла или поражения роговицы. У этих людей развиваются болезни сердца или почек в более позднем возрасте.
Признаки прогрессирующего вовлечения органов включают в себя:
- Почечная дисфункция. Прогрессирующее снижение почечной функции связано с прогрессирующим накоплением GL-3/Gb3 в почках, особенно в эндотелиальных клетках, клетках гладких мышц и подоцитах. Имеются гистологические свидетельства этого накопления и последующего клеточного и сосудистого повреждения почечной ткани, начиная с детства и юности у мужчин и женщин с классическим 1 типом заболевания. У мужчин с 1 типом снижение обычно начинается с вовлечения подоцитов и микроальбуминурии, приводящей к откровенной протеинурии (наличие в моче белка), увеличивающейся потере функции (снижению скорости клубочковой фильтрации или СКФ), все это приводит к почечной недостаточности и необходимости диализа или трансплантации, как правило, в 35-45 лет У мужчин с 2 том поражение почек обычно происходит в четвертое десятилетие или позже, но у некоторых пациентов не развивается почечная недостаточность. Участие почек у гетерозиготных женщин с 1 типом более вариабельно. Только около 10-15% женщин с типом 1 развивают почечную недостаточность. Не ясно, у какого процента женщин с 2 типом развивается почечная недостаточность, если таковая имеется.
- Болезни сердца. Отложение GL-3/Gb3 можно обнаружить во всех тканях сердца, включая клапаны, кардиомиоциты, нервы и коронарные артерии. Болезнь сердца включает в себя увеличение сердца, обычно гипертрофию левого желудочка (ГЛЖ), приводящую к гипертрофической кардиомиопатии (ГКМП), нарушениям ритма (аритмии) и сердечной недостаточности. ГЛЖ встречается примерно у 20% мужчин и женщин со средним возрастом диагноза в возрасте 20-40 лет среди мужчин с 1 типом и в конце 30-40 лет среди гетерозиготных женщин с 1 типом. Раннее поражение сердца у мужчин обычно включает аритмии и митральную недостаточность в возрасте 20 лет, после чего ГЛЖ приводит к ГКМП. У мужчин с поздним началом 2 типа развиваются те же сердечные проявления, что и у мужчин с 1 типом, но в более старшем возрасте.
- Цереброваскулярные осложнения. В результате прогрессирующего отложения GL-3/Gb3 в сердце, приводящего к фибрилляции предсердий, и в мелких кровеносных сосудах головного мозга, около 7% мужчин и 4% женщин с болезнью Фабри, особенно с фенотипом 1 типа испытывают ишемические или геморрагические инсульты, возникающие обычно в четвертом десятилетии жизни или позже.
- Нарушения дыхания. Накопление гликосфинголипидов и последующий фиброз могут вызвать интерстициальное заболевание легких. Патологические изменения и ремоделирование тканей могут включать в себя как альвеолы, так и бронхиальное дерево, что приводит к рестриктивному заболеванию легких, обструктивному заболеванию дыхательных путей или сочетанию обструктивного и рестриктивного заболевания. Респираторные симптомы у этих пациентов могут возникать независимо от сердечно-сосудистых заболеваний.
- Другие осложнения: потеря слуха, шум в ушах, головокружение, возможно, из-за отложения GL-3/Gb3 в вестибулярных структурах и/или слуховой нейропатии, обычно регистрируются у взрослых пациентов и, хотя они не опасны для жизни, способствуют бремени болезней и негативно влияют на качество жизни. Сообщалось о депрессии, и часть этих случаев, особенно мужчины с классическим 1 типом, были классифицированы как имеющие тяжелую депрессию.
Причины болезни Фабри
Болезнь Фабри вызвана изменениями (мутациями) в альфа-галактозидазе А (GLA) ген, расположенный на Х-хромосоме. Хромосомы находятся в ядре всех клеток. Они несут генетические характеристики каждого человека в тысячах специфических сегментов, называемых «генами», которые охватывают длину хромосом. Каждый из этих генов имеет определенную функцию в организме.
Человеческие хромосомы организованы в пары, пронумерованные от 1 до 22, с 23-й парой Х- и Y-хромосом у мужчин и двумя Х-хромосомами у женщин. Люди наследуют одну хромосому в каждой паре от каждого родителя. Следовательно, при Х-связанных расстройствах, включая болезнь Фабри, признаки заболевания на Х-хромосоме могут маскироваться или уменьшаться у женщин с помощью нормального гена на другой Х-хромосоме.
Если быть точнее, поскольку мужчинам и женщинам требуется только одна функционирующая Х-хромосома, одна из Х-хромосом в каждой клетке женщины, по сути, «выключена», как правило, случайным образом (случайная инактивация Х-хромосомы).
Это означает, что при Х-связанных расстройствах некоторые клетки будут иметь Х-хромосому с активированным мутировавшим геном «Фабри», в то время как другие будут иметь Х-хромосому с активированным нормальным геном. Следовательно, при болезни Фабри симптомы и степень поражения органов зависят от процента клеток в ткани/органе, где Х-хромосома с мутацией гена GLA активна, но с отсутствием или заметно сниженной функцией, что частично объясняет, почему тяжесть заболевания у женщин более вариабельна, чем у мужчин. Поскольку у мужчин есть только одна Х-хромосома, если у мужчины есть Х-хромосома с мутировавшим геном GLA, он будет страдать от этого расстройства. Следовательно, мужчины с классическим 1 и 2 типом с поздним началом заболевания Фабри поражаются более равномерно, в то время как симптомы у женщин вследствие случайной X-инактивации могут варьировать от бессимптомного или такого же серьезного поражения, как у мужчин.
Мужчины с Х-сцепленной болезнью Фабри передают мутацию гена GLA всем своим дочерям, которые являются гетерозиготами, но никогда сыновьям. Женщины гетерозиготы имеют 50% риск передачи заболевания каждому из своих детей, как дочерям, так и сыновьям, при каждой беременности.
Ген GLA обычно инструктирует клетки организма вырабатывать фермент α-Gal А, который расщепляет накапливающиеся гликолипиды (GL-3/Gb3) в лизосомах клетки. Болезнь Фабри вызвана мутациями в гене GLA. Имеется более 965 зарегистрированных мутаций в гене GLA, которые ответственны за болезнь Фабри, вызывающая фенотипы типа 1 или 2. Таким образом, тяжесть и спектр симптомов могут варьироваться среди людей в зависимости от GLA мутации в их семье. Некоторые мутации заметно изменяют фермент так, что он практически не имеет активности. Эти мутации вызывают классический подтип 1 типа, в то время как другие мутации приводят к небольшому количеству остаточной активности фермента и подтипу с поздним началом 2 типа.
Признаки и симптомы болезни Фабри развиваются из-за отсутствия или заметно недостаточной активности фермента α-Gal А. Пациенты с классическим фенотипом типа 1, которые не имеют или имеют очень низкие уровни активности (менее 3% от нормы), накапливают гликолипидное вещество GL-3/Gb-3 (и родственные гликолипиды) в большинстве тканей организма, особенно малых кровеносных сосудах и некоторых клетках сердца и почек. Пациенты с более поздним фенотипом типа 2 имеют остаточную активность фермента (3-15% от средней нормальной активности), также накапливают GL-3/Gb3, но в меньшей степени и с более медленной скоростью. Они имеют тенденцию к несколько менее тяжелой форме заболевания, но у мужчин с подтипом типа 2 в конечном итоге развивается тяжелая болезнь сердца и/или почечная недостаточность. Есть также мутации в ген GLA, которые являются доброкачественными и не вызывают болезнь Фабри.
Затронутые группы населения
Болезнь Фабри — это редкое панэтническое заболевание, которое означает, что оно встречается во всех расовых и этнических группах населения, поражающих мужчин и женщин. По оценкам, классическая болезнь Фабри типа 1 поражает примерно одного из 40 000 мужчин. Фенотип типа 2 с более поздним началом встречается чаще, чем фенотип типа 1, в 3-10 раз, и в некоторых популяциях он встречается так часто, как приблизительно 1 на 1500-4000 мужчин. Данные, полученные в результате скрининговых исследований новорожденных в США, показывают, что заболеваемость болезнью Фабри варьирует в разных географических регионах.
Диагностика
У болезни много разных симптомов, и врачи разных специальностей могут заподозрить ее возникновение. Он часто распознается неврологом после инсульта у молодого человека, нефрологом после заболевания почек или дерматологом на основании характерных изменений кожи.
Когда известно, что заболевание возникает в семье, оно может быть выявлено на ранней стадии у других членов семьи, особенно у мужчин, как только появляются первые симптомы.
Болезнь Фабри может быть обнаружена еще у плода при пренатальном исследовании.
Обычно пациент проходит серию тестов для оценки возможного повреждения различных органов. Для инсульта выполняются визуальные тесты, такие как КТ и МРТ. В случае болезни сердца, ЭКГ, эхокардиография и иногда коронарная ангиография. Заболевание почек проявляется вначале протеинурией, гематурией, а затем повышенным уровнем креатинина в крови.
Наличие описанных выше симптомов со стороны различных органов, особенно если они типичные, позволяет с уверенностью распознать заболевание. Однако для некоторых диагнозов необходимо наличие дефицита или отсутствия альфа-галактозидазы А в крови, лейкоцитах или культивируемых фибробластах.
Лечение болезни Фабри
Болезнь Фабри вызывает полиорганную дисфункцию, и пациенты нуждаются в комплексном междисциплинарном плане лечения, который индивидуально разработан и включает в себя специальные методы лечения, направленные на ненормальное накопление субстрата, и вспомогательные методы лечения, направленные на повреждение конечного органа.
Ферментозаместительная терапия (ФЗТ) является краеугольным камнем лечения болезни Фабри, а синтетический фермент, полученный с помощью технологии рекомбинантной ДНК, вводится внутривенно. Доступны две формы рекомбинантного фермента, альфа-агалсидаза (Реплагал®) и бета-агальсидаза (Фабразим®). Фабразим является единственным ФЗТ, одобренным Управлением по контролю за продуктами и лекарствами в 2003 году. Реплагал и Фабразим доступны в Европе, в России и других регионах мира. ФЗТ заменяет недостающий фермент и уменьшает накопленные гликолипиды в клетках по всему организму. Двойные слепые, плацебо-контролируемые клинические испытания фазы 3 и 4 продемонстрировали безопасность и эффективность Фабразима.
Показано, что ФЗТ замедляет или предотвращает снижение функции почек, особенно если оно начато на ранней стадии до прогрессирующего поражения почек, улучшает нейропатическую боль и непереносимость тепла. Раннее начало ФЗТ важно, особенно у мужчин с классическим типом 1. Введение ФЗТ в настоящее время рекомендуется для мужчин с классическим 1 типом с клиническими проявлениями в любом возрасте или без симптомов к 15 годам. Рекомбинантные «биоподобные» ферменты доступны в некоторых странах, включая Корею и Японию. Несколько других рекомбинантных ферментных препаратов находятся в клинической разработке.
Также для лечения взрослых с болезнью Фабри в ЕС (2017) и в США (2018) была одобрена пероральная терапия Мигаластатом. Препарат является средством, который может связывать, стабилизировать и усиливать остаточную ферментативную активность определенных миссенс-мутаций. Клинические исследования продемонстрировали эффективность этого подхода. Будущие исследования определят клиническую и биохимическую эффективность специфических миссенс-мутаций с остаточной активностью.
Дополнительные методы лечения включают низкие суточные дозы дифенилгидантоина, карбамазепина или нейронтина, чтобы помочь в управлении акропарестезией. Другие поздние осложнения (например, почечная недостаточность или проблемы с сердцем) следует лечить симптоматически после консультации с врачом, который имеет опыт ухода за пациентами с болезнью Фабри. В случаях, когда почечная недостаточность прогрессировала могут быть необходимы гемодиализ и почечная трансплантация.
Прогноз
Ожидаемая продолжительность жизни мужчин с болезнью Фабри сегодня составляет около 58 лет, а продолжительность жизни женщин с болезнью Фабри составляет чуть более 75 лет. Наиболее распространенной причиной смерти обоих полов является отказ почек и сердечно-сосудистое заболевание.
https://emedicine.medscape.com/article/1952086-overview
https://ghr.nlm.nih.gov/condition/fabry-disease
https:/rarediseases.org/rare-diseases/fabry-disease/