Что такое синдром Протея?
Синдром Протея — редкое заболевание, характеризующееся разрастанием различных тканей тела. Причина заболевания — мозаичная форма мутации гена AKT1. Непропорциональный, асимметричный разрастание происходит в виде мозаики (т.е. случайного «пятнистого» рисунка пораженных и непораженных участков).
Пострадавшие люди могут испытывать широкий спектр осложнений, которые могут включать прогрессирующие пороки развития скелета, доброкачественные и злокачественные опухоли, пороки развития кровеносных сосудов, буллезную болезнь легких и определенные поражения кожи. У некоторых людей могут развиться опасные для жизни состояния, связанные с нарушением свертываемости крови, включая тромбоз глубоких вен и тромбоэмболию легочной артерии.
Признаки и симптомы
Синдром Протея может поражать кости и соединительную ткань, жировые ткани, кожу, центральную нервную систему и внутренние органы. Специфические симптомы и степень тяжести сильно различаются от пациента к пациенту. У некоторых пациентов наблюдаются лишь несколько легких симптомов, что затрудняет диагностику.
Большинство пораженных людей рождаются без каких-либо заметных симптомов. У некоторых пациентов может быть очевиден разрастание мозга при рождении. Чрезмерный рост обычно начинается между 6-18 месяцами. Конкретные пораженные участки тела сильно различаются от пациента к пациенту. Кости, соединительная ткань и жир являются наиболее часто поражаемыми тканями в организме.
Избыточный рост, связанный с синдромом Протея, является нерегулярным, непропорциональным и может поражать одну сторону тела, например, только одну ступню. Может возникнуть чрезмерный рост костей (гиперостоз), поражающий череп, длинные кости рук и ног, а также ступни и кисти. Чрезмерный рост при болезни обычно серьезен и деформирует кости до неузнаваемости. Может быть поражен позвоночник, что приведет к сколиозу — состоянию, при котором позвоночник аномально искривлен. Прогрессирующее разрастание костной ткани в конечном итоге влияет на суставы, ограничивая диапазон движений. В конечном итоге пораженный сустав может значительно перерасти и стать неподвижным.
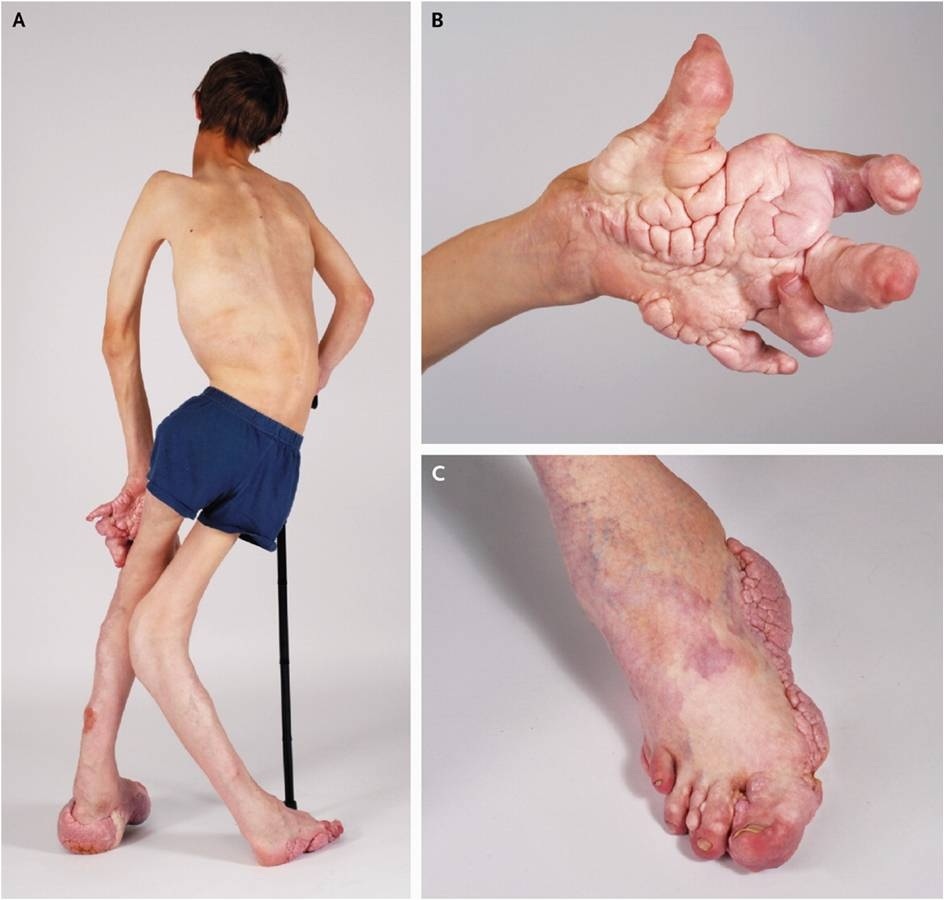
В детстве у больных могут развиться патологические состояния кожи, включая локализованные участки сильного избыточного жирового роста, особенно поражающие живот или руки и ноги. В некоторых случаях могут развиваться доброкачественные опухоли, состоящие из жировой ткани (липомы). Помимо чрезмерного разрастания жировой ткани, у некоторых больных развиваются атрофия жировой ткани, особенно в области груди.
У пораженных детей также может развиться приподнятое, бородавчатое поражение (эпидермальный невус), обычно грубое и темно-коричневого или коричневато-черного цвета. Эпидермальный невус может присутствовать при рождении. Может возникнуть еще одно поражение кожи, известное как соединительнотканные невусы мозговой оболочки. Это медленнорастущее поражение чаще всего встречается на ступнях и реже на руках. Он отсутствует при рождении и состоит из утолщенной аномально плотной подкожной клетчатки. На коже могут образоваться глубокие бороздки.
При синдроме Протея часто встречаются различные пороки развития кровеносных сосудов (сосудистые мальформации). Могут быть поражены капилляры, вены и лимфатические сосуды. Капилляры — крошечные кровеносные сосуды, соединяющие артерии и вены. Вены — кровеносные сосуды, по которым кровь поступает к сердцу. Лимфатические сосуды являются частью лимфатической системы, циркулирующей сети сосудов, протоков и узлов, которые фильтруют и распределяют определенную богатую белком жидкость (лимфу) и клетки крови по всему телу.
Люди с синдромом Протея могут подвергаться риску образования тромбов в ногах — состояния, известного как тромбоз глубоких вен нижних конечностей. Ноги могут стать болезненными и опухшими, а кровеносные сосуды ног могут заметно увеличиться. В некоторых случаях часть тромба может отсоединится и подняться по кровотоку в легкие, где может вызвать тромбоэмболию легочной артерии. Тромбоэмболия легочной артерии — застревание сгустка внутри легочной артерии, которое потенциально может вызвать одышку, внезапную боль в груди, истощение или опасные для жизни осложнения, такие как высокое кровяное давление в легочной артерии (легочная гипертензия).
При синдроме Протея могут наблюдаться дополнительные признаки, включая аномальное увеличение определенных внутренних органов, таких как селезенка, тимус, толстая кишка и другие ткани.
У больных также есть предрасположенность к развитию самых разных опухолей, большинство из которых являются доброкачественными. Опухоли, наиболее часто связанные с заболеванием, — это двусторонние цистаденомы яичников, группа редких опухолей слюнных желез, известных как мономорфные аденомы и менингиомы.
Менее распространенные проявления синдрома включают пороки развития центральной нервной системы, например, чрезмерный рост половины мозга (гемимегаленцефалия). У некоторых пациентов может присутствовать умственная отсталость, также сообщалось о эпилепсии. Люди с этими аномалиями могут также иметь отчетливые черты лица, включая удлиненное лицо (см. фото выше), наклонные вниз складки век (глазные щели), опущенные веки (птоз), низкую переносицу, широкие ноздри и длинную узкую голову (долихоцефалия). Причина ассоциации неврологических и лицевых аномалий неизвестна.
У некоторых людей с синдромом Протея может развиться кистозная болезнь легких, заболевания почек или мочевыводящих путей, а также патологии глаз, такие как косоглазие или доброкачественные кисты или опухоли глазных яблок (эпибульбарные кисты или дермоиды).
Причины
Синдром Протея вызывается мутацией гена, регулирующего рост, который называется AKT1, который возникает после оплодотворения эмбриона (соматическая мутация). У больных есть некоторые клетки с нормальной копией этого регуляторного гена и некоторые клетки с аномальным геном (мозаицизм). Разнообразие симптомов, связанных с заболеванием, отчасти связано с соотношением здоровых и аномальных (патологических) клеток. Когда все клетки имеют патологический ген, это состояние несовместимо с жизнью. Исследователи полагают, что эта соматическая мутация происходит случайно без видимой причины (спорадически).
Некоторые исследователи отнесли подгруппу пациентов с синдромом Протея к мутации гена PTEN, расположенного на хромосоме 10. Это привело к путанице больных. Другие исследователи считают, что эти пациенты, хотя в некоторых отношениях похожи на больных с синдромом Протея, их состояние не соответствуют определенным диагностическим критериям. Этот так называемый синдром Протея представляет собой совершенно другое заболевание. Ни у кого из больных с определенным подтвержденным синдрома Протея не было обнаружено наличия мутации гена PTEN.
Затронутые группы населения
Синдром Протея — крайне редкое заболевание. В медицинской литературе описано около 200 пациентов, и, похоже, что заболевание затрагивает людей всех этнических и расовых групп. Тем не менее, исследователи с большим опытом в лечении синдрома Протея изучили этих пациентов и определили, что только менее 100 человек соответствовали строгим диагностическим критериям заболевания
Поскольку диагностика заболевания настолько сложна, некоторым людям диагноз может быть не поставлен, в то время как другим диагноз может быть поставлен неправильно, когда у них вместо этого заболевания есть другое состояние. Поэтому определить истинную частоту расстройства в общей популяции крайне сложно.
Синдром Протея поражает мужчин несколько чаще, чем женщин. Впервые об этом было сообщено в медицинской литературе в 1979 году. Теперь исследователи полагают, что Джозеф Меррик, о жизни которого был снят фильм «Человек-слон», страдал синдромом Протея, а не нейрофиброматозом, как считалось ранее.
Близкие по симптома расстройства
Симптомы следующих заболеваний могут быть похожи на симптомы синдрома Протея. Сравнения могут быть полезны для дифференциальной диагностики.
- Синдром гемигиперплазии-множественного липоматоза — редкое заболевание, характеризующееся развитием множественных доброкачественных опухолей, состоящих из жировой ткани (липомы) и аномального увеличения одной стороны или структуры тела (гемигиперплазия), что приводит к неравномерному (асимметричному) росту. Гемигиперплазия может указывать на асимметрию только между одной конечностью и другой или между одной половиной тела и другой. Гемигиперплазия может быть умеренно прогрессирующей. Причиной заболевания обычно является соматическая мутация гена PIK3CA.
- Энцефалокраниокожный липоматоз — чрезвычайно редкое заболевание, характеризующееся аномалиями глаз, аномалиями кожи, включая опухоли, состоящие из жировой ткани (липомы), поражающие кожу головы и центральную нервную систему, и поражения кожи, состоящие из неправильно развитой соединительной ткани (соединительнотканные невусы). Конкретные симптомы сильно различаются от пациента к пациенту. У некоторых людей нормальный интеллект, у других может быть умственная отсталость. У некоторых людей были зарегистрированы эпилепсия и порэнцефальные кисты. Также могут присутствовать дополнительные симптомы. Хотя ранее считалось, что энцефалокраниокожный липоматоз представляет собой форму синдрома Протея, ограниченную головой и шеей, недавно исследователи определили более конкретные критерии болезни, и, похоже, что это заболевание отличается от синдрома Протея. У ряда пациентов с этим расстройством имеются мозаичные мутации в гене, называемом FGFR1.
- Синдром Клиппеля-Треноне — редкое заболевание, которое присутствует при рождении (врожденное), характеризуется наличием капиллярной сосудистой мальформации (винное пятно или пламенеющий невус) на коже руки или ноги, возникающей в связи с чрезмерным ростом (гипертрофией) мягких тканей и кости этой ноги и/или руки (конечности) и варикозное расширение вен (венозные мальформации). У некоторых пораженных людей также обнаруживается лимфатическая аномалия пораженной конечности. У людей с этим заболеванием такая гипертрофия обычно поражает одну конечность или одну сторону тела (гемигипертрофия). Симптомы и признаки, связанные с расстройством, могут варьироваться по диапазону и степени тяжести от пациента к пациенту. Синдром Клиппеля-Треноне обычно вызывается мозаичной мутацией PIK3CA и является частью спектра избыточного роста, связанного с PIK3CA.
- Синдром Маффуччи — редкое генетическое заболевание, характеризующееся доброкачественными разрастаниями хрящей (энхондромами), деформациями скелета и темно-красными участками кожи неправильной формы, возникающими в результате доброкачественных новообразований на коже (кожных покровах), состоящих из масс кровеносных сосудов (гемангиомы). Энхондромы чаще всего встречаются в определенных костях (фалангах) кистей и стоп. Скелетные пороки развития могут включать непропорциональную длину ног и/или аномальное искривление позвоночника из стороны в сторону (сколиоз). У многих людей кости легко ломаются. У большинства людей гемангиомы появляются при рождении или в раннем детстве и могут прогрессировать. Синдром Маффуччи наследуется по аутосомно-доминантному признаку.
Диагностика
Диагностика синдрома Протея проводится с использованием опубликованных клинических диагностических критериев и молекулярных тестов. Подтверждение диагноза может быть трудным, а интерпретация клинических диагностических критериев неоднозначна. Идентификация причинной мутации гена AKT1 может позволить молекулярная диагностика, хотя это тоже может быть сложной задачей. Изменение гена редко присутствует в крови, и поэтому диагностическое тестирование ДНК обычно должно выполняться на биопсиях пораженных тканей.
Другие диагностические методы, которые могут использоваться при оценке, могут включать простую рентгенографию, компьютерную томографию (КТ) для выявления поражений черепа, компьютерную томографию высокого разрешения для выявления кист легких и магнитно-резонансную томографию (МРТ) головного мозга, живота, таза и конечностей. Для обнаружения новообразований мошонки или яичников и для оценки тромбоза глубоких вен используется ультразвуковое исследование.
Стандартные методы лечения
Лечение синдрома Протея направлено на устранение конкретных симптомов, которые проявляются у каждого человека. Как правило, для того, чтобы попытаться контролировать быстрое перерастание, связанное с синдромом Протея, необходимы многочисленные ортопедические процедуры. Хирургическое вмешательство может быть необходимо, когда избыточный рост нарушает функцию суставов, вызывает сколиоз или угловые деформации. Может быть показана операция по уменьшению заросших тканей или частей тела. Эпифизиоз (удаление или аблация пластинок роста в костях) может быть особенно полезен для профилактики или лечения скелетного избыточного роста при синдроме.
Пациенты с синдромом Протея, подвергающиеся хирургическим вмешательствам, должны находиться под тщательным наблюдением, поскольку операция может предрасполагать пострадавших к тромбозу глубоких вен. При хирургическом вмешательстве следует рассмотреть вопрос о профилактике сгустков крови для предотвращения образования тромбов (антитромботическая профилактика) или лечении тромбов после их образования.
Пациентам и их семьям рекомендуется генетическое консультирование. Другое лечение симптоматическое и поддерживающее.
Прогноз
Прогноз варьируется в зависимости от тяжести осложнений.