Что такое несовершенный остеогенез?
Несовершенный остеогенез (сокр. НО или также называемая «несовершенное костеобразование», болезнь «хрустального человека», болезнь Лобштейна-Вролика) — это группа редких заболеваний, поражающих соединительную ткань и характеризующихся чрезвычайно хрупкими костями, которые легко ломаются, часто без видимой причины.
Специфические симптомы и физические признаки, связанные с заболеванием, сильно различаются от случая к случаю. Тяжесть несовершенного остеогенеза также сильно различается, даже среди членов одной семьи. НО может быть легким заболеванием или привести к тяжелым осложнениям. Выделены четыре основных типа НО. НО I типа — наиболее частая и легкая форма заболевания. НО II типа является наиболее тяжелым заболеванием.
В большинстве случаев различные формы несовершенного остеогенеза наследуются по аутосомно-доминантному признаку.
Признаки и симптомы
При всех типах несовершенного остеогенеза сопутствующие симптомы сильно различаются от случая к случаю, даже внутри семьи. Некоторые больные люди могут не иметь переломов костей или испытывать лишь некоторые из них; у других пострадавших могут быть множественные переломы. НО может варьироваться от легкого заболевания с небольшими симптомами до тяжелого, изнурительного расстройства. Возраст возникновения переломов также варьируется от случая к случаю.
— Остеогенез I типа.
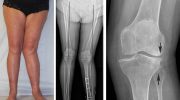
Остеогенез I типа — наиболее распространенная и обычно самая легкая форма НО. В большинстве случаев болезнь характеризуется множественными переломами костей, которые обычно происходят в детстве и в период полового созревания. Переломы обычно начинаются, когда больной ребенок начинает ходить; переломы в новорожденном (неонатальном) периоде встречаются редко. После полового созревания частота переломов обычно снижается. Повторные переломы могут привести к незначительной деформации костей рук и ног (например, искривлению большеберцовой и бедренной костей).
Отличительной чертой, связанной с НО I типа, является изменение цвета белков глаз на голубоватый оттенок (голубая склера). В некоторых случаях у людей с НО I типа могут развиваться патологии среднего и/или внутреннего уха, способствующие или приводящие к нарушению слуха (например, кондуктивная и/или нейросенсорная потеря слуха). Чаще всего потеря слуха возникает в третьем десятилетии жизни; однако это может произойти уже во второй декаде или уже в седьмой декаде.
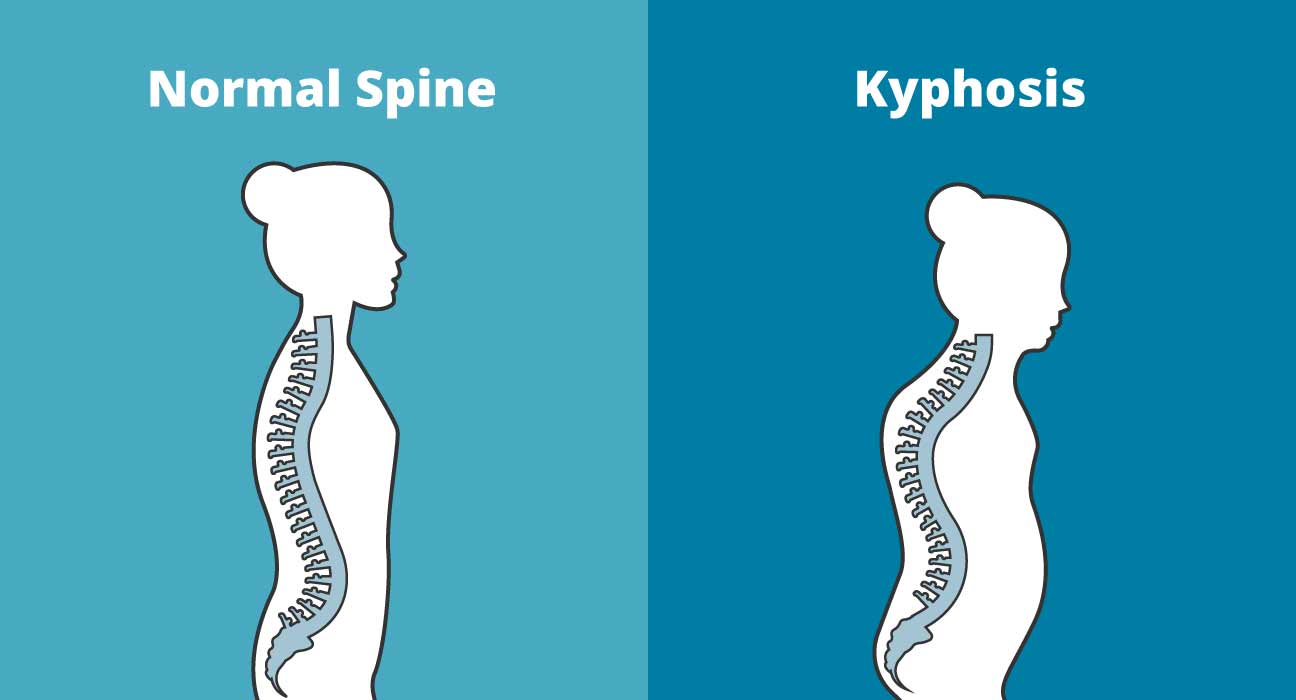
Люди с НО I типа могут иметь треугольную форму лица и аномально большую голову (макроцефалия). Примерно в 50% случаев люди с несовершенным остеогенезом I типа будут испытывать дефицит роста после рождения (послеродовой период), что приводит к небольшому росту (например, больные люди будут ниже ростом, чем здоровые члены семьи). Приблизительно у 20 процентов взрослых с НО I типа развивается аномальное искривление позвоночника в стороны или спереди назад (сколиоз или кифоз).
Дополнительные симптомы, связанные с НО I типа, включают свободные (гиперрастяжимые) суставы, низкий мышечный тонус (гипотония) и тонкую кожу, на которой легко появляются синяки.
Некоторые исследователи полагают, что существует подгруппа НО I типа, в которой пациенты имеют патологии зубов в дополнение к вышеупомянутым особенностям.
— Остеогенез II типа.
НО II типа — наиболее тяжелый тип несовершенного остеогенеза. У заболевших младенцев часто возникают опасные для жизни осложнения при рождении или вскоре после рождения. Младенцы с НО типа II имеют низкий вес при рождении, аномально короткие руки и ноги (конечности) и голубоватое изменение цвета белков глаз (голубые склеры). Кроме того, у пораженных младенцев могут быть чрезвычайно хрупкие кости и многочисленные переломы при рождении. Ребра и длинные кости ног пораженных младенцев часто деформируются.
Младенцы с НО типа II часто имеют недоразвитые легкие и аномально малую верхнюю часть грудной клетки, что может привести к опасной для жизни дыхательной недостаточности. В некоторых случаях у пораженных младенцев может наблюдаться застойная сердечная недостаточность.
У младенцев с НО типа II может быть маленький узкий нос; маленькая челюсть (микрогнатия); и аномально мягкая верхняя часть черепа (свод черепа) с аномально большими мягкими пятнами. У заболевших младенцев также может быть ненормально тонкая, хрупкая кожа и низкий мышечный тонус (гипотония).
Несовершенный остеогенез II типа подразделяется на три подгруппы (A, B и C) на основании небольших различий в формировании кости, видимых только на рентгеновских снимках (рентгенологические особенности).
— Остеогенез III типа.
НО типа III характеризуется чрезвычайно хрупкими костями, множественными переломами и деформированными костями. Множественные переломы часто возникают при рождении. Переломы и уродства различных костей (чаще всего ребер и длинных костей) могут усугубляться (прогрессирующая деформация) по мере взросления младенцев и детей.
Прогрессирующая деформация различных костей может привести к низкому росту, искривлению позвоночника в стороны и спереди назад (сколиоз и кифоз), а также к деформации области задней части черепа (затылочная кость) и верхней части позвоночника (базилярная инвагинация). В некоторых случаях у пораженных людей может развиться легочная недостаточность и проблемы с дыханием. В тяжелых случаях прогрессирующая деформация костей может привести к тому, что пациенты будут нуждаться в инвалидных колясках.
У младенцев с НО типа III при рождении может наблюдаться легкое изменение цвета белков глаз в голубой цвет. В большинстве случаев голубоватый оттенок исчезает в течение первого года жизни. Пораженные младенцы могут иметь треугольный вид лица из-за аномально выступающего лба и аномально маленькой челюсти. В некоторых случаях также могут присутствовать нарушение слуха и хрупкие, обесцвеченные зубы (несовершенный дентиногенез).
— Остеогенез IV типа.
У людей с НО типа IV хрупкие кости, которые часто легко ломаются. Переломы чаще встречаются до полового созревания. Пораженные люди имеют пороки развития костей от легкой до умеренной, и их длина обычно ниже среднего. У больных может развиться искривление позвоночника в стороны и спереди назад (сколиоз и кифоз).
Лица с НО 4 типа могут иметь треугольную форму лица. В большинстве случаев белки глаз (склеры) в младенчестве нормальные или бледно-голубые. По мере того как больной ребенок взрослеет, бледно-голубое изменение цвета склеры исчезает. У больных также может наблюдаться проблемы со слухом и несовершенный дентиногенез.
— Неклассифицированный несовершенный остеогенез.
В медицинской литературе сообщалось о многих случаях людей с аномалиями костей, характерными для несовершенного остеогенеза. Однако эти случаи имеют дополнительные симптомы, которые не позволяют отнести их к одному из четырех основных типов НО. Исследователи предположили, что эти случаи могут быть подгруппами одного из четырех основных типов НО, дополнительными типами НО (например, НО типа V) или отдельными расстройствами.
Причины
В большинстве случаев несовершенный остеогенез типов I, II и IV наследуется как аутосомно-доминантные признаки. Большинство случаев НО типа II возникает без семейного анамнеза заболевания, а является результатом спонтанного генетического изменения (т.е. новой мутации).
Человеческие черты, в том числе классические генетические заболевания, являются продуктом взаимодействия двух генов, одного от отца, а другого от матери. При доминантных расстройствах единственная копия гена болезни (полученная от матери или отца) будет выражаться «доминирующей» над другим нормальным геном и приводить к появлению болезни. Риск передачи заболевания от пораженного родителя к потомству составляет 50 процентов при каждой беременности независимо от пола родившегося ребенка. Риск одинаков для каждой беременности.
Редкие подгруппы НО типов II и III могут быть унаследованы как аутосомно-рецессивные генетические признаки. При рецессивных расстройствах это состояние не возникает, если индивидуум не наследует один и тот же дефектный ген одного и того же признака от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, он будет носителем болезни, но обычно не бессимптомным. Риск передачи болезни детям пары, оба из которых являются носителями рецессивного заболевания, составляет 25% при каждой беременности. Риск иметь ребенка-носителя, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальную копию гена от обоих родителей, и будут генетически нормальными (для этого конкретного признака) составляет 25%. Риск одинаков для мужчин и женщин.
Исследователи определили, что большинство типов несовершенного остеогенеза вызвано нарушением или изменениями (мутациями) одного из двух генов (COL1A1 или COL1A2). Эти гены несут инструкции по производству коллагена 1 типа. Коллаген — это основной белок костей и соединительной ткани, включая кожу, сухожилия и склеры.
Ген COL1A1 расположен на длинном плече (q) хромосомы 17 (17q21.31-q22). Хромосомы находятся в ядрах всех клеток организма. Они несут в себе генетические характеристики каждого человека. Пары человеческих хромосом пронумерованы от 1 до 22, с неравной 23-й парой хромосом X и Y у мужчин и двумя хромосомами X у женщин. У каждой хромосомы есть короткое плечо, обозначенное буквой «p», и длинное плечо, обозначенное буквой «q». Хромосомы далее подразделяются на пронумерованные полосы. Например, «хромосома 17q21.31-q22» относится к полосам 21.31-22 на длинном плече хромосомы 17.
Ген COL1A2 расположен на длинном плече (q) хромосомы 7 (7q22.1).
В 2006 году исследователи из Национального института здоровья (англ. National Institutes of Health (NIH)) обнаружили, что ранее необъяснимая фатальная форма несовершенного остеогенеза является результатом генетического дефекта в другом гене, известном как CRTAP. Ген CRTAP кодирует (т.е. содержит информацию) белок, связанный с хрящом. Эти мутации связаны с рецессивной формой заболевания. Открытие этого гена было описано в статье, опубликованной 28 декабря 2006 года в Медицинском журнале Новой Англии.
Затронутые группы населения
Несовершенный остеогенез поражает мужчин и женщин в равном количестве. Точное количество людей с НО в России (распространенность) неизвестно. НО I типа, по оценкам, встречается у 1 из 30 000 живорожденных. НО типа II встречается у 1 из 60 000 живорожденных. Общая распространенность всех типов НО оценивается в 0,5 на 10 000 человек.
Близкие по симптомам расстройства
Симптомы следующих нарушений могут быть аналогичны симптомам несовершенного остеогенеза. Сравнения могут быть полезны для дифференциальной диагностики:
- Ахондроплазия — это наследственное заболевание, характеризующееся аномально короткими руками и ногами и низким ростом (карликовость коротких конечностей), аномальными чертами лица и/или пороками развития скелета. Характерные черты лица могут включать аномально большую голову (макроцефалия), необычно выступающий лоб, низкую переносицу и/или недоразвитие средней части лица (гипоплазия средней части лица). Скелетные пороки развития могут включать необычно короткие пальцы рук и ног (брахидактилия), аномально увеличенное искривление позвоночника назад (лордоз), сгибание ног наружу и/или сужение (стеноз) позвоночника. Дополнительные патология могут включать ограниченное разгибание локтей и бедер, снижение мышечного тонуса (гипотония) и/или частые инфекции среднего уха (средний отит). В большинстве случаев, ахондроплазия возникает случайно, без видимой причины (спорадически).
- Гипофосфатазия — редкое заболевание, характеризующееся дефектным укреплением (минерализацией) костей, что приводит к ослаблению костей. Длинные кости рук и ног могут быть слишком толстыми, короткими и искривленными. У пораженных младенцев также может быть аномально маленькая голова (микроцефалия). Многие взрослые с гипофосфатазией имеют низкий рост. Гипофосфатазия передается по наследству как аутосомно-рецессивный признак.
- Пикнодизостоз — редкое заболевание, характеризующееся повышенной плотностью костей (остеосклероз), низким ростом, недоразвитой нижней челюстью и заболеваниями зубов. У больных часто есть хрупкие кости и они могут быть подвержены стрессовым переломам. Пикнодизостоз передается по наследству как аутосомно-рецессивный признак.
- Остеопетроз — редкое заболевание, характеризующееся повышенной плотностью костей, ломкостью костей и, в некоторых случаях, заболеваниями скелета. Хотя симптомы могут изначально не проявляться у людей с легкими формами этого расстройства, тривиальные травмы могут вызывать переломы костей из-за патологий кости. Взрослый тип остеопетроза более мягкий, чем детский злокачественный и промежуточный остеопетроз, и может быть диагностирован только в подростковом или взрослом возрасте, когда впервые появляются симптомы. Более серьезные осложнения возникают при младенческом злокачественном и промежуточном остеопетрозе. Остеопетроз может передаваться по наследству по аутосомно-рецессивному или доминантному признаку.
- Синдром несовершенного остеогенеза — это термин, используемый для описания группы заболеваний, характеризующихся патологиями костей (например, хрупкими костями и множественными переломами), аналогичных тем, которые обнаруживаются при основных четырех типах НО. Однако эти расстройства имеют связанные особенности, которые отличают их от основных четырех типов несовершенного остеогенеза. Эти так называемые синдромы несовершенного остеогенеза крайне редки, и в медицинской литературе описано всего несколько случаев каждого синдрома. Описанный ниже синдром Коула-Карпентера является синдромом несовершенного остеогенеза.
- Синдром Коула-Карпентера — чрезвычайно редкое заболевание, характеризующееся чрезвычайно хрупкими костями и множественными переломами, подобными тем, которые наблюдаются при НО. Кроме того, у больных младенцев может наблюдаться голубой цвет склер, преждевременное закрытие фиброзных суставов (краниальные швы) между некоторыми костями черепа (краниосиностоз), аномально выступающий лоб, аномально маленькая челюсть и выпячивание глаза (проптоз). У пораженных младенцев также может наблюдаться задержка роста. В медицинской литературе описано около 5 случаев этого расстройства. Синдром Коула-Карпентера наследуется по аутосомно-доминантному признаку.
Диагностика
Диагноз несовершенного остеогенеза ставится на основании тщательной клинической оценки, подробного анамнеза пациента, выявления характерных симптомов и различных специализированных тестов. Для определения наличия аномалий коллагена могут быть выполнены хирургическое удаление и микроскопическое исследование (биопсия) кожи. Может быть взят образец крови и протестирован для выявления генетической мутации, вызывающей НО.
В некоторых случаях диагноз НО может быть поставлен до рождения (пренатально) на основании специализированных тестов, таких как УЗИ, амниоцентез и/или биопсии ворсин хориона. Ультразвуковые исследования могут выявить характерные признаки, такие как переломы или искривление длинных костей. Во время амниоцентеза образец жидкости, окружающей развивающийся плод, удаляется и исследуется. Во время взятия пробы ворсин хориона образец ткани берется из части плаценты. Хромосомные исследования, проведенные на этом образце жидкости или ткани, могут выявить генетическую мутацию, вызывающую остеогенез.
Стандартные методы лечения
Лечение несовершенного остеогенеза направлено на устранение конкретных симптомов, которые проявляются у каждого человека. Лечение направлено на предотвращение симптомов, поддержание индивидуальной подвижности и укрепление костей и мышц.
Программы упражнений и физиотерапии доказали свою эффективность в укреплении мышц, повышении способности выдерживать нагрузку и снижении склонности к переломам. Физиотерапия в воде (гидротерапия) также оказалась полезной, поскольку движение в воде снижает вероятность перелома. Лица с НО должны проконсультироваться со своими врачами и физиотерапевтами, чтобы определить безопасную и подходящую программу упражнений.
Часто для лечения лиц с НО используется процедура, при которой в длинные кости хирургическим путем помещаются металлические стержни для предотвращения переломов. В качестве защитных приспособлений пластиковые скобы заменяют гипсовые повязки, поскольку они обеспечивают большую свободу движений и могут использоваться в воде. Надувные костюмы могут обеспечить дополнительную защиту, особенно очень маленьким детям.
Хирургическое вмешательство может оказаться необходимым для людей с серьезной деформацией костей позвоночника или базилярной инвагинацией. Для исправления различных аномалий зубов могут потребоваться стоматологические процедуры. Пациентов, особенно взрослых, следует обследовать на предмет нарушения слуха, часто связанного с НО. Другие методы лечения являются симптоматическими и поддерживающими. Семьям, в которых встречается это заболевание, рекомендуется генетическое консультирование.
Прогноз
Прогноз людей с несовершенным остеогенезом сильно различаются в зависимости от типа заболевания. Большинство детей, рожденных с НО I типа, до взрослого возраста живут нормальной здоровой жизнью. Менее тяжелые симптомы не влияют на продолжительность жизни. Большинство смертей, связанных с НО, наступает в результате дыхательной недостаточности из-за слабости легких.