Что такое синдром Маршалла?
Синдром Маршалла является редким аутосомно-доминантным генетическим заболеванием, вызванным мутациями в гене COL11A1. Основные симптомы синдрома Маршалла у детей могут включать характерное изменение лица с уплощенным носовым мостом и ноздрями, смотрящие вверх, широко расставленные глаза, близорукость, катаракту и потерю слуха. Люди с расстройством также могут иметь низкий рост.
Некоторые исследователи утверждают, что синдром Маршалла представляет собой разновидность синдрома Стиклера; однако это остается спорным вопросом.
Признаки и симптомы
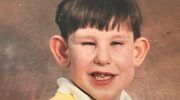
Дети и взрослые с синдромом Маршалла имеют характерную плоскую затонувшую среднюю часть лица с уплощенным носовым мостом, ноздри, смотрящие вверх, и широкое пространство между глазами (гипертелоризм). Верхняя часть черепа, похожая на купол, толще, чем обычно, а в черепе можно обнаружить отложения кальция. Лобные пазухи могут отсутствовать. Глазные дефекты у пациентов с синдромом Маршалла — это близорукость, заболевание глаза, при котором хрусталик теряет четкость (катаракта), и большое пространство между глазами, из-за которого глазные яблоки кажутся больше, чем обычно. Потеря слуха может варьироваться от легкой до тяжелой; искажение звука является следствием повреждения нерва (нейросенсорной). Другие симптомы, проявляющиеся у некоторых пациентов с синдромом Маршалла:
- косоглазие (эзотропия);
- состояние, при котором линия зрения на одном глазу выше, чем на другом (гипертропия);
- отслоение сетчатки;
- глаукома;
- выступающие верхние резцы (зубы);
- меньшая по сравнению с нормой или отсутствующая носовая кость.
Причины
Синдром Маршалла — редкое аутосомно-доминантное генетическое заболевание, вызванное мутациями в гене полипептида альфа-1 коллагена (COL11A1), расположенном на хромосоме 1p21.1. Обычно мутации, вызывающие синдром Маршалла, представляют собой мутации в месте сращивания, включающие вставки базовой пары или делеции интрона 50. Доминантные генетические нарушения возникают, когда необходима только одна копия ненормального гена, чтобы вызвать конкретное заболевание. Аномальный ген может быть унаследован от любого из родителей или может быть результатом новой мутации (изменения гена) у пострадавшего человека. Риск передачи ненормального гена от пострадавшего родителя к потомству составляет 50% при каждой беременности.
Сообщалось об одной саудовской семье с двумя сыновьями с синдромом Маршалла с гомозиготными миссенс-мутациями COL11A1. В этой семье было беспокойство о возможном аутосомно-рецессивном наследовании, так как у каждого родителя была одна миссенс-мутация с замещением глицина, и у этих родителей был низкий рост, утолщенный череп и легкая потеря слуха при нормальном офтальмологическом обследовании и у них не было диагноза синдрома Стиклера или Маршалла. Рецессивные генетические нарушения возникают, когда ребенок наследует две ненормальные копии гена, по одной от каждого здорового родителя. Возможно, в этой семье оба родителя слабо подвержены синдрому Стиклера, и в этой ситуации наследование будет названо двойной доминантой. В любой ситуации риск рецидива будет 25% с каждой беременностью. Риск одинаков для мужчин и женщин.
Затронутые группы населения
Из-за редкости заболевания очень мало демографических данных. В медицинской литературе по всему миру зарегистрировано менее 100 случаев этого синдрома. Некоторые случаи, вероятно, не диагностированы из-за высокой стоимости генетического тестирования. Известно, что синдром Маршалла проявляется в младенчестве или раннем детстве, а серьезные симптомы, такие как потеря слуха и катаракта, проявляются в возрасте до 10 лет. Синдром Маршалла поражает мужчин и женщин в равных количествах.
Близкие по симптомам расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Маршалла. Сравнения могут быть полезны для дифференциальной диагностики:
- Врожденная спондилоэпифизарная дисплазия — редкое генетическое заболевание, характеризующееся дефицитом роста еще до рождения (внутриутробным), пороками развития позвоночника и/или аномалиями, влияющими на глаза. По мере старения больных дефицит роста в конечном итоге приводит к низкому росту (карликовости), частично из-за непропорционально короткой шеи и туловища, и деформации бедра, при которой бедренная кость наклонена к центру тела. В большинстве случаев у пострадавших могут наблюдаться снижение мышечного тонуса (гипотония), аномальная кривизна спереди и сзади и поперек (кифоз и сколиоз), аномальная кривизна позвоночника внутрь (поясничный лордоз) и/или необычные выпячивание грудины, состояние, известное как воронкообразная грудь. Пострадавшие люди также имеют нарушения, влияющие на глаза, включая близорукость и, приблизительно в 50 процентах случаев отслоение богатой нервом мембраны, выстилающей глаз (сетчатки). Врожденная спондилоэпифизарная дисплазия наследуется как аутосомно-доминантный признак, также связанный с мутациями, делециями и дупликациями гена COL2A1, с аутосомно-доминантным наследованием.
- Врожденный сифилис — хроническое инфекционное заболевание, вызываемое спирохетой (бледной трепонемой), приобретенной плодом в матке. Симптомы заболевания могут проявляться не ранее, чем через несколько недель или месяцев после рождения, а в некоторых случаях они могут проявляться годами. Врожденный сифилис передается ребенку от матери, которая приобрела заболевание до или во время беременности. Симптомы раннего врожденного сифилиса включают лихорадку, проблемы с кожей и низкий вес при рождении. При позднем врожденном сифилисе симптомы заболевания обычно проявляются только в возрасте от двух до пяти лет. Симптомами позднего врожденного сифилиса могут быть боль в костях, колючие верхние центральные резцы (зубы), помутнение зрения, боль в глазах и нечувствительность к свету, седлообразный нос, костный выступ лба, короткая верхняя челюстная кость и глухота. В редких случаях заболевание может оставаться скрытым в течение многих лет с симптомами, не диагностируемыми до зрелого возраста.
- Синдром Стиклера относится к группе нарушений соединительной ткани, которые затрагивают различные системы органов организма, такие как глаза, скелет, внутреннее ухо и/или голову и лицо. Соединительная ткань, которая является материалом между клетками тела, придающая ткани форму и силу, имеется по всему телу. Соединительная ткань состоит из белка, известного как коллаген, в организме которого есть несколько различных разновидностей. Синдром Стиклера часто поражает соединительную ткань глаза, особенно внутреннюю часть глазного яблока (стекловидное тело), специализированную ткань, которая служит буфером или подушкой для костей в суставах (хрящ) и на концах костей, которые составляют суставы тела (эпифиз). Пять различных форм синдрома Стиклера были определены в медицинской литературе на основании местоположения мутированного гена, клинических симптомов и характера наследования. Тип I с мембранозным стекловидным телом, вызванным мутациями в гене COL2A1; тип II вызванная мутациями в гене COL11A1; тип III является неглазным типом, вызванным мутациями в COL11A2, тип IV вызывает дегенерацию стекловидного тела с прогрессирующим сжижением и имеет аутосомно-рецессивное наследование с мутациями в COL9A1, тип V имеет те же клинические симптомы, что и типы I и II, но расположение мутация еще не найдена.
- Синдром Стиклера типа II вызывается мутациями в том же гене COL11A1, что и синдром Маршалла. Некоторые исследователи считают, что эти два расстройства являются одинаковыми или разными выражениями одного и того же расстройства. Другие считают, что эти два расстройства различны. Недавние исследования показывают, что мутации в гене COL11A1, связанные с синдромом Маршалла, являются сплайсинговыми мутациями в экзонах в С-концевых областях COL11A1 с «горячей точкой» в экзоне 50.
- Синдром Вагнера является очень редким генетическим заболеванием, наследуемый как аутосомно-доминантный признак и вызванный мутацией в гене CSPG2 в хромосоме 5q13-14. Ген CSPG2 кодирует версикан, структурный компонент стекловидного тела. Это было обнаружено в исследовании одной швейцарской семье. К глазным проблемам данного синдрома относятся витреоретинальная дегенерация и катаракта. Отслойки сетчатки встречаются крайне редко. Внеглазных проблем не обнаруживалось.
Стандартные методы лечения
Пластическая хирургия может улучшить седловидный нос при синдроме Маршалла у детей. Другие хирургические процедуры используются для удаления линз глаз, пораженных катарактой, после чего в качестве замены используются искусственные хрусталики. Впоследствии контактные линзы могут помочь улучшить резкость зрения. Лазерные методы используются для ослабления любого материала, например роговицы или капсулы хрусталика, которая может прилипнуть к линзе.
Использование слухового аппарата может быть полезным в некоторых случаях. Для пострадавших и их семей рекомендуется генетическая консультация. Другое лечение является симптоматическим и поддерживающим.
Прогноз
Информация о средней продолжительности жизни человека с синдромом Маршалла ограничена. В одной из статей описывается семья, в которой много затронутых родственников в нескольких поколениях, и упоминаются лица, страдающие синдромом Маршалла, которым за 30 и 40 лет. Имеются также конкретные сообщения о лицах, которые предположительно имеют синдром Маршалла и хорошо себя чувствуют в возрасте 29 и 35 лет.
Из медицинской литературы следует, что лица с синдромом Маршалла могут иметь относительно нормальную продолжительность жизни. Ожидается, что основные признаки синдрома Маршалла не будут представлять угрозы для жизни, хотя степень тяжести симптомов может варьироваться у пострадавших лиц. Лишь в одной статье медицинской литературы говорится, что продолжительность жизни может быть сокращена в связи с наличием одного состояния; авторы высказывают предположение, что проблемы с дыханием, связанные с синдромом Маршалла, могут привести к сокращению продолжительности жизни. Однако в настоящее время проблемы с дыханием не рассматриваются в качестве признака этого заболевания, и ни в одной другой статье эта проблема не упоминается.