Общая информация
Синдром Мартина-Белла (синдром ломкой X-хромосомы) — это наследственное заболевание, характеризующееся умеренной умственной отсталостью у мальчиков и девочек. У пораженных мальчиков иногда присутствуют другие отличительные физические особенности, включая большую голову, длинное лицо, выступающий лоб и подбородок, выступающие уши и большие яички, но эти признаки развиваются со временем и могут не проявляться до полового созревания. Моторные и языковые задержки обычно присутствуют, но со временем они становятся все более очевидными. Поведенческие отклонения, включая аутистическое поведение, являются общими для обеих полов.
Синдром ломкой X-хромосомы был обнаружен во всех основных этнических группах и расах и вызван аномалией (мутацией) в гене FMR1. FMR1 — это ген, расположенный на Х-хромосоме, продуцирующем белок, называемый FMRP, необходимый для правильного функционирования клеток. Синдром стал называться как синдром ломкой X-хромосомы, поскольку у некоторых людей с этим расстройством был обнаружен сегмент их Х-хромосомы, который казался сломанным (хотя и не полностью отсоединенным). Позже стало известно, что ген FMR1 расположен именно там, где Х-хромосома кажется «ломкой» у пораженных людей.
Хромосомы, которые присутствуют в ядре клеток человека, несут генетическую информацию каждого человека. Клетки человеческого тела обычно имеют 46 хромосом. Пары человеческих хромосом пронумерованы от 1 до 22, а половые хромосомы обозначены X и Y. У мужчин есть одна Х и одна Y-хромосома, а у женщин две Х-хромосомы. Каждая хромосома имеет короткое плечо, обозначенное «p», и длинное плечо, обозначенное «q». Хромосомы далее подразделяются на множество пронумерованных полос. Например, «хромосома Xq27.3» относится к полосе 27,3 на длинном плече Х-хромосомы, где расположен ген FMR1. Пронумерованные полосы указывают местоположение тысяч генов, присутствующих в каждой хромосоме.
Х-связанные доминантные расстройства, такие как синдром ломкой X-хромосомы, вызваны ненормальным геном, расположенным на Х-хромосоме. Женщины с аномальным геном могут быть затронуты этим расстройством. Мужчины, как правило, более серьезно болеют, чем женщины.
Именно отсутствие или серьезное уменьшение белка, вырабатываемого геном FMR1, FMRP, вызывает синдром Мартина-Белла. Мутация гена FMR1 вызывает потерю или уменьшение FMRP. Почти все затронутые индивидуумы имеют нестабильность в гене, что приводит к увеличению числа копий части гена, называемой тринуклеотидным повтором цитозин-гуанин-гуани (ЦГГ). При наличии более 200 повторов происходят аномальные химические изменения в FMR1, называемые метилированием. Расширение области повторов ЦГГ до более чем 200 повторов, сопровождаемое метилированием гена, называемого «полной мутацией», вызывает потерю FMRP, приводя к синдрому Мартина-Белла. Болезнь чаще встречается у мужчин и у них он протекает более тяжело.
Мутации в FMR1 необычны по сравнению с мутациями, обнаруженными в других генах. Некоторые люди носят от 55 до 200 ЦГГ-повторов, называемых «премутациями», обычно без симптомов, связанных с синдромом Мартина-Белла. Тем не менее, эти люди подвержены риску рождения детей или внуков с синдромом Мартина-Белла, а также риску возникновения двух расстройств, возникающих во взрослом возрасте синдрома хрупкого X-связанного тремора/атаксии и первичной яичниковой недостаточности. Эти заболевания были названы расстройствами, связанными с FMR1. (Пожалуйста, ознакомьтесь с разделами «Причины и близкие по симптомам расстройства» в этой статье, чтобы получить более подробные объяснения относительно предварительных оценок и краткого описания этих нарушений, связанных с FMR1).
Признаки и симптомы
Синдром Мартина-Белла характеризуется умеренной умственной отсталостью у затронутых мужчин и женщин. Физические особенности у затронутых мужчин изменчивы и могут не быть очевидными до полового созревания. Эти симптомы могут включать в себя:
- большую голову;
- длинное лицо;
- выступающий лоб и подбородок;
- выступающие уши;
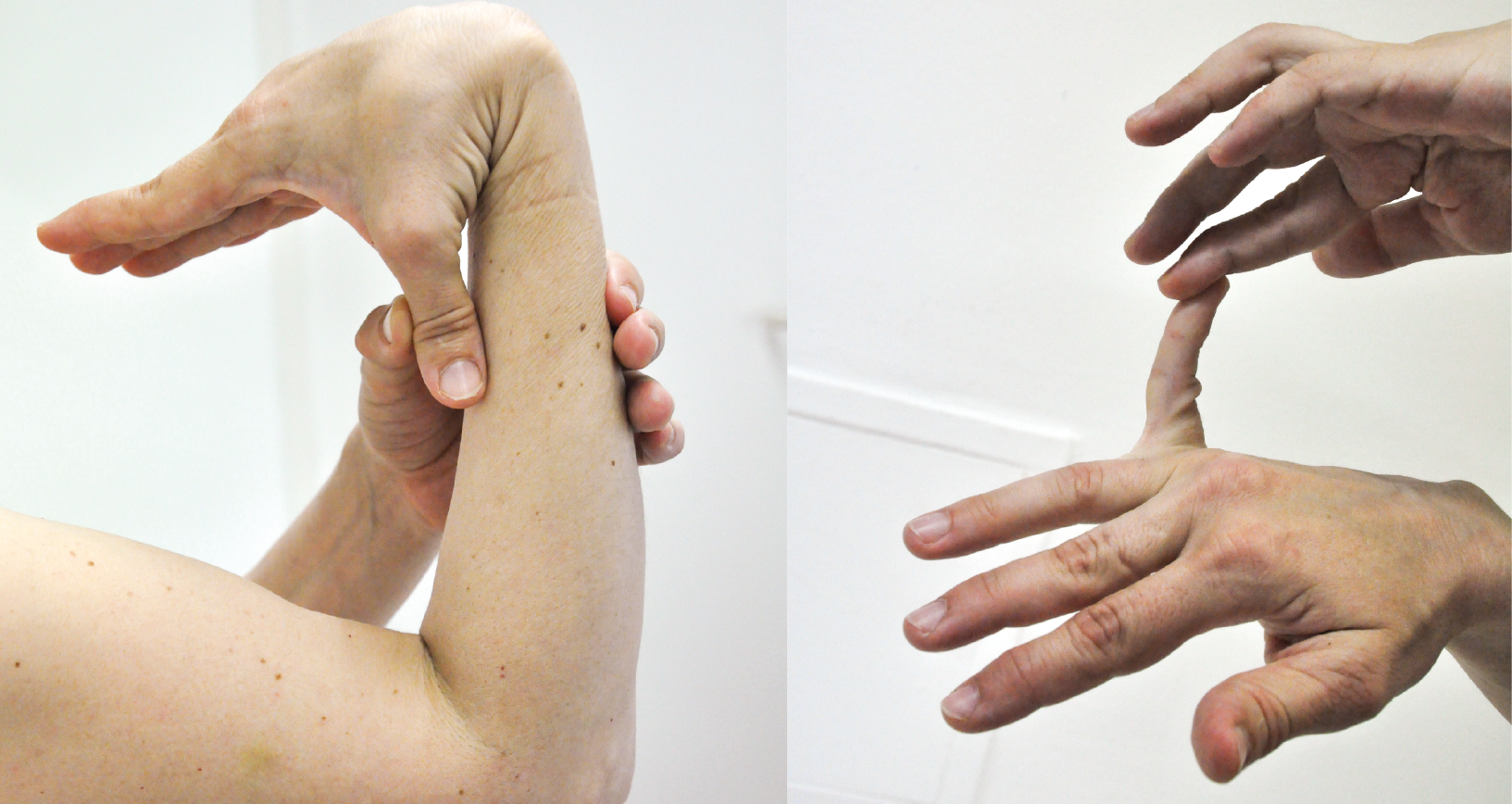
- рыхлые суставы;
- крупные яички.
Другие симптомы могут включать:
- плоскостопие;
- частые ушные инфекции;
- низкий тонус мышц;
- длинное узкое лицо;
- высокое сводчатое нёбо;
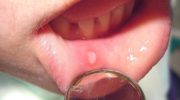
- проблемы с зубами;
- косоглазие;
- проблемы с сердцем, включая пролапс митрального клапана.
У некоторых пациентов также могут наблюдаться задержки в развитии моторики, гиперактивность, проблемы с поведением, ходьба на носочках и/или эпизодические судороги. Также часто встречаются аутичное поведение, например, плохой зрительный контакт, хлопание руками и/или самостимулирующее поведение. Моторные и языковые задержки обычно присутствуют, но со временем они становятся еще более очевидными.
Причины
Как упомянуто выше, синдром Мартина-Белла (синдром ломкой X-хромосомы) вызван мутацией в гене FMR1, расположенном на Х-хромосоме в Xq27.3. Люди с синдромом ломкой X-хромосомы почти всегда имеют (более чем в 99% случаев) полную мутацию гена FMR1, что означает, что у них более 200 повторов ЦГГ и аномальное метилирование гена. Метилирование — это химическое изменение ДНК, которая несет генетический код гена, а аномальное метилирование, связанное с синдромом Мартина-Белла, приводит к тому, что ген не способен продуцировать FMRP, белок, вырабатываемый геном FMR1, необходимый для нормального развития.
В редких случаях у некоторых пациентов с синдромом частично или полностью отсутствует ген FMR1 из-за делеции ДНК на Х-хромосоме, где находится FMR1, и имеют этот синдром из-за того, что их клетки не вырабатывают FMRP. Было обнаружено, что у ультра-редких пациентов с заболеванием наблюдается мутация в одной основе ДНК (так называемые точечные мутации), приводящая к отсутствию или дефекту FMRP. FMRP участвует в создании связей между нейронами (нервными клетками) в мозге. Отсутствие или серьезное снижение этого белка приводит к симптомам синдрома Мартина-Белла.
Премутации (предварительные мутации) имеют 55-200 ЦГГ-повторов и потенциально нестабильны. Лица, имеющие премутацию гена FMR1, не имеют синдрома Мартина-Белла, но взрослые подвержены риску возникновения синдрома хрупкого X-связанного тремора/атаксии и первичной яичниковой недостаточности.
При передаче из поколения в поколение предварительные мутации могут быть нестабильными и становиться полными мутациями, но риск нестабильности различен в зависимости от того, передает ли премутацию женский или мужской пол. Женщины с премутацией гена FMR1 подвергаются риску родить ребенка с синдромом Мартина-Белла, поскольку количество повторов ЦГГ может увеличиваться при передаче гена следующему поколению. Чем больше копий ЦГГ в премутации, тем больше вероятность того, что они увеличатся и станут полной мутацией, вызывающей синдром у потомства.
Когда мужчины с премутацией размножаются, их дети мужского пола не рискуют унаследовать премутацию, поскольку отцы не передают Х-хромосому своим сыновьям. Напротив, женщины, чьи отцы имеют премутацию, всегда наследуют ее, и, таким образом, внуки мужчин с премутацией подвержены риску возникновения синдрома ломкой X-хромосомы. Поскольку премутация относительно стабильна при передаче от отца к дочери, дочери почти никогда не подвержены синдрому ломкой X-хромосомы. Однако их дети подвергаются повышенному риску, поскольку премутация может быть нестабильной при передаче ее следующему поколению.
Нормальные гены FMR1 имеют примерно 5-44 ЦГГ-повторов, и это число остается стабильным из поколения в поколение. Иногда, у некоторых людей с 45-54 повторениями будет некоторая незначительная нестабильность, так что у этих людей будет несколько (или меньше) повторов, чем у их родителей. Число повторов FMR1 между 45 и 54 называется «промежуточным» или «серой зоной», но эта незначительная нестабильность не приводит к появлению каких-либо симптомов синдрома Мартина-Белла или расстройств, связанных с FMR1. Наличие промежуточного числа повторов ЦГГ все еще считается находящимся в нормальном диапазоне числа повторений.
Затронутые группы населения
Синдром Мартина-Белла поражает примерно 1 из 4000 мужчин и 1 из 6000-8000 женщин; т.е., заболевание затрагивает примерно вдвое больше мужчин, чем женщин. Тем не менее, примерно в четыре раза больше женщин являются носителями измененного гена, чем мужчины (1:250 женщин и 1:1000 мужчин). Синдром ломкой X-хромосомы был обнаружен во всех основных этнических группах и расах.
Близкие по симптомам расстройства
Премутации гена FMR1 связаны с двумя другими расстройствами, и эти состояния называются расстройствами, связанными с FMR1. Не у всех людей с премутацией будут развиваться расстройства, связанные с FMR1, но наличие премутации увеличивает риск их развития.
- Синдром хрупкого X-связанного тремора/атаксии характеризуется прогрессирующими аномалиями движения у взрослых (атаксией) и ритмичными непроизвольными движениями (тремором), которые поражают в основном мужчин. Люди с этим заболеванием имеют премутацию в гене FMR1 (55-200 повторов ЦГГ). Диагностика этого синдрома может быть осложнена его сходством с другими поздними взрослыми расстройствами, такими как болезнь Паркинсона.
- Связанная с FMR1 первичная яичниковая недостаточность (ПЯН) определяется как менопауза в возрасте до 40 лет у женщин с премутацией в гене FMR1 (55-200 повторов ЦГГ). Риск возникновения ПЯН у носительниц премутации составляет примерно 21%. Женщины с ПЯН неизвестной причины имеют риск 1/50 быть носительницами премутации в гене FMR1.
Некоторые симптомы следующих расстройств также могут быть похожи на симптомы синдрома Мартина-Белла. Сравнения могут быть полезны для дифференциальной диагностики:
- Синдром Фрейкса встречается редко и вызывается аномальным геном FMR2, расположенным на Х-хромосоме очень близко к месту гена FMR1. Нормальный ген FMR2 содержит 6-35 копий CCG, а люди с расстройством имеют более 200 копий CCG в гене FMR2. Эффект генов FMR2 с 35-200 копиями CCG еще не был определен, вероятно, потому что расстройство имеет умеренную клиническую картину. Общие симптомы синдрома Фрейкса включают умеренную умственную отсталость, возможные задержки в развитии.
- Синдром Ренпеннинга является одним из хромосомных Х-связанных расстройств умственной отсталости, который затрагивает мальчиков почти исключая девочек. Очень редко у девочек присутствует этот синдром. Он характеризуется серьезной умственной отсталостью, невысоким ростом, маленькой головой (микроцефалия) и маленькими яичками. Задержка развития проявляется при синдроме Ренпеннинга на ранней стадии, когда дети учатся ходить в возрасте 2–3 лет и могут произносить простые слова в возрасте 3–4 лет. Хотя больной мальчик может казаться физически нормальным, его окружность головы и рост будут находиться в нижних пределах нормы. После полового созревания яички будут меньше, чем обычно. Диагностика очень сложна, особенно если в семье только один человек с ограниченными умственными возможностями. Диагноз должен основываться на доказательствах наследования как признака Х-хромосомы и определения того, что пораженный ген находится на коротком плече (в Xp11.1-p11.4) Х-хромосомы.
Диагностика
Более 99% людей с синдромом Мартина-Белла имеют полную мутацию (более 200 повторов ЦГГ и аномальное метилирование) в гене FMR1. Для определения количества ЦГГ-повторов в гене FMR1 используется молекулярно-генетическое тестирование, а для определения статуса метилирования гена FMR1 часто используется тестирование с целью выявления расширенной области ЦГГ.
Хромосомный анализ с использованием специальных методов для индукции ломких участков в хромосомах когда-то использовался для диагностики синдрома Мартина-Белла, но больше не используется с этой целью. Синдром ломкой X-хромосомы — это второе название, данное этому состоянию, поскольку у некоторых пораженных людей Х-хромосома выглядела так, как будто она «сломалась» и была скреплена малейшей из связей. Этот метод больше не используется в диагностике этого синдрома, потому что он менее точный и дорогой, чем молекулярные методы.
Стандартные методы лечения
Существует множество способов лечения синдрома Мартина-Белла, которые могут улучшить жизнь пострадавших людей и их семей. Они включают в себя специальное образование, обучение речи, профессиональную и сенсорную интеграцию, а также программы изменения поведения. Благодаря образовательным усилиям, терапии и поддержке все люди с синдромом Мартина-Белла могут добиться прогресса. Дополнительное лечение может зависеть от конкретных симптомов больного. Для пострадавших людей и их семей также рекомендуется генетическое консультирование.
В США и во всем мире существует множество клиник, где лечат синдром ломкой X-хромосомы. Эти клиники специализируются на лечении, терапии и поддержке людей с синдромом и могут помочь родителям в выборе лекарств для устранения специфических симптомов. Скорее всего, появятся новые лекарства для лечения больных, и специализированные клиники могут помочь родителям с актуальной информацией.
Прогноз
Продолжительность жизни людей с синдромом ломкой X-хромосомы, как правило, нормальная. Многие пострадавшие люди ведут активный образ жизни и имеют хорошее здоровье. Некоторые люди более склонны к ряду заболеваний, например, ушным инфекциям и/или эпилептическим припадкам. Регулярные медицинские осмотры и осведомленность о повышенных рисках для здоровья могут улучшить прогнозы больных.