Что такое синдром Пейтца–Егерса?
Синдром Пейтца-Егерса (сокр. СПЕ) — это аутосомно-доминантное генетическое заболевание, поражающее примерно от 1/50 000 до 1/200 000 человек. Симптомы обычно появляются в течение первого десятилетия жизни и начинаются с веснушчатых пятен на темной коже (меланоцитарные пятна) вокруг рта, глаз, ноздрей, пальцев, а также внутри рта и вокруг ануса. Множественные доброкачественные полипы, называемые гамартомами, также начинают расти в желудочно-кишечном тракте больных людей примерно в этом возрасте. Эти полипы расположены по всему желудочно-кишечному тракту и могут вызывать тошноту, рвоту, боль в животе, кишечную непроходимость и ректальное кровотечение. Для удаления полипов (полипэктомии) может потребоваться абдоминальная хирургия или эндоскопические процедуры, чтобы предотвратить связанные с полипами осложнения, такие как инвагинация кишечника. Больные c синдромом Пейтца–Егерса имеют повышенный риск рака кишечника и других видов рака. Для раннего выявления полипов и рака необходимы частые медицинские осмотры и анализы.
Синдром Пейтца-Егерса является частью гетерогенной группы заболеваний, известных как синдромы гамартоматозного полипоза, которые включают рост множественных полипов в желудочно-кишечном тракте больных. Впервые он был описан у пары сестер-близнецов с темными пигментными пятнами на губах и слизистой оболочке рта доктором Дж. Т. Коннором в 1895 году. Эти симптомы были отнесены к семейному синдрому в 1921 году, когда доктор Ян Пейтц описал четырех пострадавших братьев и сестер. Затем этот синдром был определен как отдельное расстройство доктором Гарольдом Егерсом в 1949 году, когда он описал 10 случаев, и впоследствии был назван синдромом Пейтца–Егерса в 1954 году доктором Андре Брювером. Ген, вызывающий расстройство (STK-11/LKB1), был идентифицирован в 1998 году и позволяет раннее выявить заболевание и провести скрининг членов семьи.
Признаки и симптомы
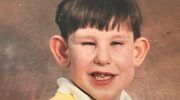
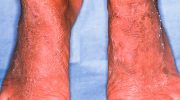
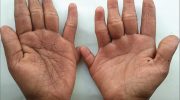
Синдром Пейтца–Егерса характеризуется ростом множественных доброкачественных полипов, называемых гамартомами, на слизистой оболочке желудочно-кишечного тракта и пятнами от темно-синего до темно-коричневого цвета с веснушками (меланоцитарные пятна) вокруг рта, глаз, ноздрей, пальцев, слизистой оболочки полости рта и ануса. Эти меланоцитарные пятна могут появиться уже на первом году жизни и присутствуют у большинства пораженных детей в возрасте до пяти лет. Они имеют тенденцию исчезать с возрастом и могут полностью исчезнуть в период полового созревания или взрослой жизни, хотя они, как правило, сохраняются в слизистой оболочке полости рта.
Полипы также начинают расти в первые годы жизни, но связанные с ними симптомы обычно возникают в возрасте от 10 до 30 лет. Около половины пациентов с расстройством должны подвергнуться операции к 18 годам из-за осложнений, связанных с полипами. Чаще всего полипы развиваются в тонкой кишке (в частности, в тощей кишке), но также могут возникать в желудке и толстой кишке. В редких случаях полипы могут расти за пределами желудочно-кишечного тракта и поражать мочеточники, мочевой пузырь, легкие, бронхи и желчный пузырь. Полипы желудочно-кишечного тракта могут вызывать боль в животе, рвоту, диарею, кишечную непроходимость и ректальное кровотечение, что может привести к анемии. Также они могут спровоцировать сворачивание кишечника внутрь себя (инвагинацию), что может привести к сильной боли в животе и экстренной операции.
Лица с синдромом Пейтца-Егерса подвержены высокому риску развития рака желудочно-кишечного тракта и других видов рака, включая рак груди, шейки матки, рак матки, рак поджелудочной железы и рак легких. Риск развития рака на протяжении всей жизни у пораженных людей может достигать 93%. Люди, у которых развивается рак, обычно заболевают примерно на пятом десятилетии жизни (возраст 40-49 лет). У пораженных женщин повышен риск доброкачественной опухоли яичников, симптомы которой могут включать нерегулярные или тяжелые менструации или раннее половое созревание. Обычно в возрасте до 20 лет у пораженных мужчин может развиться опухоль в яичках, называемая карциномой из клеток Сертоли, которая выделяет эстроген и может привести к увеличению груди (гинекомастия).
Причины
Синдром Пейтца-Егерса — аутосомно-доминантное генетическое заболевание, вызванное мутациями в гене STK11/LKB1. Доминирующие генетические нарушения возникают, когда для появления болезни необходима только одна копия аномального (патологического) гена. Патологический ген может быть унаследован от любого из родителей или может быть результатом новой мутации у пораженного человека. Риск передачи аномального гена от пораженного родителя к потомству составляет 50% при каждой беременности. Риск одинаков для мужчин и женщин.
Приблизительно у 60-78% людей с расстройством есть больные родственники. Приблизительно у 80-94% пациентов с синдромом Пейтца-Егерса выявлена мутация в гене STK11, что означает, что в заболевание могут быть вовлечены и другие гены. Сообщалось о более чем 200 болезнетворных (патогенных) мутациях, и считается, что пенетрантность этих мутаций составляет 100%, что означает, что у человека, несущего патогенную мутацию, обязательно разовьется болезнь.
Ген STK11 производит белок, который участвует в регуляции клеточного деления и программируемой гибели клеток (апоптоза). Он также взаимодействует с р53, основным белком подавления опухоли. Патогенные мутации в STK11 приводят либо к прекращению, либо к дисфункции продукции белка геном и неконтролируемому росту клеток, что, в свою очередь, может привести к развитию доброкачественных полипов (гамартомам) и раку.
Считается, что темные пигментные пятна (меланоцитарные пятна) вызваны воспалением и блокировкой миграции меланина из клеток, где он вырабатывается (меланоциты), в клетки, образующие самый внешний слой кожи (кератиноциты).
Затронутые группы населения
Синдром Пейтца-Егерса — редкое заболевание, которое поражает в равных количествах мужчин и женщин и может встречаться в любой расовой или этнической группе. Распространенность расстройства при рождении составляет от 1/50 000 до 1/200 000. Ограниченные данные показывают, что болезнь может быть более распространена в некоторых странах, таких как Нидерланды и Китай. Женщины подвергаются более высокому риску развития рака по сравнению с мужчинами, поскольку синдром Пейтца-Егерса увеличивает вероятность развития рака груди, яичников, шейки матки и матки.
Близкие по симптомам расстройства
Симптомы следующих заболеваний могут быть похожи на симптомы синдрома Пейтца-Егерса. Сравнения могут быть полезны для дифференциальной диагностики:
- Синдром ювенильного полипоза — это аутосомно-доминантное генетическое заболевание, характеризующееся специфическим типом гамартоматозного полипа, называемым ювенильным полипом. Большинство полипов доброкачественные, но у пораженных людей повышен риск рака толстой кишки и других видов рака.
- Синдром зубчатого полипоза — аутосомно-доминантное генетическое заболевание, характеризующееся определенным типом полипа, называемым зубчатым полипом (аденомой), расположенным в толстой кишке. Большинство полипов доброкачественные, но зубчатый полипоз связан с более частым заболеванием колоректальным раком в личном и семейном анамнезе.
- Семейный аденоматозный полипоз (САП) — редкий синдром наследственной предрасположенности к раку, характеризующийся наличием сотен и тысяч предраковых колоректальных полипов (аденоматозных полипов). Если не лечить, у пораженных людей неизбежно развивается рак толстой и/или прямой кишки в относительно молодом возрасте. САП наследуется по аутосомно-доминантному типу и вызывается мутациями в гене APC.
- Синдром Тюрко — редкое наследственное заболевание, характеризующееся ассоциацией доброкачественных новообразований (аденоматозных полипов) на слизистой оболочке желудочно-кишечного тракта с опухолями центральной нервной системы. Симптомы, связанные с образованием полипа, могут включать диарею, кровотечение из концевой части толстой кишки (прямой кишки), утомляемость, боль в животе и потерю веса. У больных также могут возникать неврологические симптомы в зависимости от типа, размера и местоположения связанной опухоли головного мозга. Исследователи считают, что синдром Тюрко является вариантом семейного аденоматозного полипоза или синдрома Линча (наследственный неполипозный колоректальный рак).
- Синдром наследственного смешанного полипоза — аутосомно-доминантное генетическое заболевание, характеризующееся развитием нескольких типов полипов (атипичные ювенильные полипы, гиперпластические полипы, зубчатые аденомы и аденоматозные полипы) в желудочно-кишечном тракте. Больные люди подвержены повышенному риску колоректального рака.
- Опухолевый синдром PTEN-гамартом представляет собой спектр заболеваний, вызванных мутациями в гене-супрессоре опухоли PTEN. Эти нарушения характеризуются множественными гамартомами, которые могут поражать различные участки тела. Признаки также включают повышенный риск определенных типов рака и нарушений развития нервной системы. Симптомы расстройства сильно различаются от человека к человеку и могут развиться в любом возрасте.
- Синдром Кронкхайта-Канады (СКК) — чрезвычайно редкое заболевание, характеризующееся различными кишечными полипами, потерей вкуса, потерей волос и проблемами роста ногтей. СКК встречается в основном у пожилого населения (средний возраст 59 лет) и преимущественно у мужчин. СКК считается приобретенным, а не наследственным заболеванием.
- Множественная эндокринная неоплазия 2 типа (МЭН 2) — редкое генетическое заболевание, характеризующееся повышенным риском развития конкретной формы рака щитовидной железы (медуллярная карцинома щитовидной железы) и доброкачественных опухолей, поражающих дополнительные железы эндокринной системы. У людей с одним конкретным типом синдрома, называемым МЭН 2B, могут развиваться доброкачественные новообразования, возникающие из нервных клеток, называемых ганглиозными клетками (ганглионевроматоз). Эти новообразования возникают в желудочно-кишечном тракте и могут вызывать вздутие живота, диарею, запор и аномально увеличенную толстую кишку (мегаколон). Больные младенцы часто не набирают вес и не растут с ожидаемой скоростью для их возраста и пола (неспособность развиваться).
- Комплекс Карни — это редкое генетическое заболевание, характеризующееся множественными доброкачественными опухолями (множественная неоплазия), чаще всего поражающими сердце, кожу и эндокринную систему, а также отклонениями в окраске кожи (пигментации), приводящими к появлению пятен на коже пораженных участков. У людей с комплексом Карни распространены доброкачественные опухоли соединительной ткани (миксомы), которые чаще всего встречаются в сердце, где они могут вызвать серьезные, угрожающие жизни осложнения, в том числе инсульт, обструкцию венозной артерии или сердечную недостаточность. При комплексе Карни потенциально могут возникать самые разные эндокринные нарушения, затрагивающие различные железы. Дополнительные опухоли включают миксомы, поражающие кожу и опухоли нервных оболочек (шванномы). Нарушения пигментации кожи включают крошечные плоские (похожие на веснушки) черные или коричневые пятна (множественные лентиго) и небольшие, синие или голубовато-черные пятна (голубые невусы).
Диагностика
Клинический диагноз синдрома Пейтца-Егерса (СПЕ) может быть поставлен при наличии любого из следующих критериев:
- Наличие как минимум двух полипов синдрома Пейтца-Егерса.
- Любое количество полипов и хотя бы один близкий родственник с диагнозом синдрома Пейтца-Егерса.
- Характерные темные пигментные пятна (меланоцитарные пятна) и хотя бы один близкий родственник с диагнозом СПЕ.
- Любое количество полипов и характерных темных пигментных пятен.
Меланоцитарные пятна можно определить при физическом осмотре. Полипы могут быть обнаружены с помощью эндоскопии, рентгенологического исследования или эндоскопии с помощью беспроводной капсулы и классифицируются как полипы синдрома Пейтца-Егерса при микроскопическом исследовании.
Генетическое тестирование для выявления болезнетворных (патогенных) мутаций в гене STK11 рекомендуется при наличии любого из следующих критериев:
- Характерные темные пигментные пятна.
- Наличие как минимум двух полипов СПЕ.
- Семейная история синдрома Пейтца-Егерса.
Генетическое тестирование особенно полезно, когда в семье уже выявлена патогенная мутация. Считается, что пенетрантность мутации гена STK11 составляет 100% (что означает, что у человека с патогенной мутацией есть 100% вероятность заболеть заболеванием), поэтому генетическое тестирование может дать точный диагноз до появления симптомов. Около половине пациентов с СПЕ диагноз ставится с помощью генетического тестирования до появления симптомов.
Стандартные методы лечения
В первую очередь больным следует получить консультацию клинического генетика или генетического консультанта. Поскольку от синдрома Пейтца-Егерса нет лекарства, лечение в основном направлено на наблюдение и контроль над симптомами. После первоначального диагноза людям старше 8 лет или имеющим симптомы рекомендуется пройти эндоскопическое обследование и обследование тонкой кишки. Последнее может быть выполнено с помощью магнитно-резонансной томографии кишечника (магнитно-резонансная энтерография, МР-энтерография) или путем проглатывания капсулы, которая делает внутренние изображения из желудочно-кишечного тракта (видеокапсульная эндоскопия). Гинекологическое обследование и обследование груди также рекомендуется женщинам старше 18 лет. Мужчинам рекомендуется обследование яичек.
После первоначального обследования после постановки диагноза каждые 2–3 года следует проводить эндоскопию, колоноскопию и обследование тонкой кишки для выявления полипов и потенциальных опухолей. Женщинам рекомендуется делать ежегодно маммографию. Мужчинам УЗИ яичек можно делать раз в два года.
Поскольку синдром Пейтца-Егерса увеличивает риск рака груди, матки и яичников, больные женщины могут пройти профилактическую мастэктомию, гистерэктомию или сальпингоофорэктомию (хирургическое удаление груди, матки, маточных труб и яичников соответственно).
Полипы размером более 1 см удаляются эндоскопическими методами, чтобы избежать связанных с полипами осложнений, таких как кровотечение и инвагинация. Эти осложнения могут потребовать хирургического вмешательства. Если пациенту предстоит операция, эндоскопическое удаление полипов (полипэктомия) выполняется одновременно с операцией, чтобы снизить риск рецидива осложнений и хирургического вмешательства.
В случаях, когда темные пигментные пятна (меланоцитарные пятна) оказывают крайне негативное психологическое воздействие на пораженных людей, их можно частично удалить с помощью лазерной терапии.
Прогноз
Синдром Пейтца-Егерса (СПЕ) нельзя полностью вылечить — это пожизненное расстройство, которое может передаваться по наследству. Людям с синдромом Пейтца-Егерса необходимо часто проверяться на наличие полипов. Эти полипы могут перерасти в рак или вызвать закупорку, что может потребовать хирургического вмешательства.