Что такое синдром Вернера?
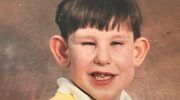
Синдром Вернера (или прогерия взрослых) — редкое прогрессирующее заболевание, которое характеризуется появлением необычно ускоренного старения (прогерии). Хотя расстройство обычно распознается к третьему или четвертому десятилетию жизни, определенные характерные признаки присутствуют, начиная с подросткового и раннего взросления.
У людей с синдромом Вернера скорость роста низкая, и рост прекращается в период полового созревания. В результате больные имеют низкий рост и низкий вес по сравнению с ростом. К 25 годам пациенты с этим заболеванием обычно испытывают раннее поседение (седину) и преждевременное выпадение волос на коже головы (алопеция). По мере прогрессирования болезни дополнительные аномалии включают потерю слоя жира под кожей (подкожной жировой ткани); сильное истощение (атрофия) мышечной ткани в определенных областях тела; и дегенеративные изменения кожи, особенно в области лица, предплечий и кистей рук, а также голеней и ступней (дистальных отделов конечностей). Из-за дегенеративных изменений, затрагивающих лицевую область, больные с синдромом Вернера могут иметь необычно выпуклые глаза, клювообразный или защемленный нос и/или другие характерные лицевые аномалии.
Синдром Вернера также может характеризоваться развитием характерного высокого голоса; глазными аномалиями, в том числе преждевременным помутнением хрусталиков глаз (двусторонняя старческая катаракта); и некоторыми эндокринными дефектами, такими как нарушение функции яичников у женщин или яичек у мужчин (гипогонадизм) или аномальное производство гормона инсулина поджелудочной железой и резистентность к действию инсулина (инсулиннезависимый сахарный диабет/диабет 2-го типа). Кроме того, у больных с синдромом Вернера может развиваться прогрессирующее утолщение и потеря эластичности стенок артерий (артериосклероз). Пораженные кровеносные сосуды обычно включают артерии, которые транспортируют богатую кислородом (насыщенную кислородом) кровь к сердечной мышце (коронарным артериям). Некоторые больные также могут быть подвержены развитию определенных доброкачественных или злокачественных опухолей. Прогрессирующий артериосклероз, злокачественные новообразования и/или связанные с ними нарушения могут привести к потенциально опасным для жизни осложнениям примерно к четвертому или пятому десятилетию жизни. Синдром Вернера наследуется по аутосомно-рецессивному типу.
Признаки и симптомы
Дети с синдромом Вернера часто выглядят необычно худыми и в позднем детстве имеют необычно медленный темп роста. Кроме того, отсутствует скачок роста, обычно наблюдаемый в подростковом возрасте. Больные достигают своего окончательного роста примерно к 13 годам. Однако взрослый рост может быть достигнут уже в возрасте 10 лет или уже в возрасте 18 лет. Вес также необычно маленький, даже по сравнению с невысоким ростом.
До 20 лет у большинства людей с синдромом Вернера наблюдается раннее поседение и обесцвечивание волос на коже головы. Примерно к 25 годам у больных может наблюдаться преждевременная потеря волос на коже головы (алопеция), а также потеря бровей и ресниц. Кроме того, волосы под мышками (волосы в подмышечных впадинах), в области лобка и на туловище могут быть необычно редкими или отсутствовать. Согласно сообщениям в медицинской литературе, выпадение волос, наблюдаемое у пациентов с синдромом Вернера, может быть вторичным по отношению к нарушению функции яичников у женщин или яичек у мужчин (гипогонадизм), эндокринного состояния, связанного с недостаточным ростом и половым развитием. И мужчины, и женщины с синдромом Вернера могут страдать от гипогонадизма. В результате у пораженных мужчин обычно необычно маленький пенис и маленькие яички. У некоторых женщин с этим заболеванием могут не развиться вторичные половые признаки (например, появление волос в подмышечных впадинах и на лобке, развитие груди, менструация) и плохо развитые половые органы. У других больных женщин менструация может быть скудной и нерегулярной. Из-за гипогонадизма большинство людей с этим заболеванием могут быть бесплодны. Однако в литературе были сообщения, подтверждающие, что некоторые больные мужчины и женщины размножались.
Помимо преждевременного поседения и выпадения волос, больные с синдромом Вернера подвержены другим прогрессирующим дегенеративным изменениям, включая
- постепенную потерю слоя жира под кожей;
- сильное истощение (атрофия) мышц рук, ног и ступней;
- преждевременная общая потеря плотности костной ткани (остеопороз), состояние, которое может вызывать или способствовать повторным переломам после незначительной травмы.
Также могут присутствовать стоматологические аномалии, включая аномальное развитие и преждевременную потерю зубов. Примерно у одной трети пациентов с синдромом Вернера также наблюдается аномальное накопление солей кальция (кальцификация) и связанное с этим затвердевание мягких тканей (например, связок, сухожилий), особенно локтей, коленей и лодыжек. К тому же, из-за прогрессирующей атрофии голосовых связок у большинства больных с этим заболеванием развивается высокий голос. В других случаях голос может быть скрипучим или необычно хриплым.
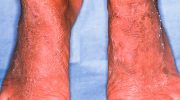
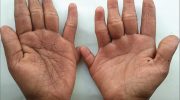
Примерно к 25 годам у больных с синдромом Вернера также развиваются прогрессирующие кожные изменения, особенно в области лица, предплечий и кистей рук, а также голеней и ступней (дистальных отделов конечностей). Например, наблюдается потеря (атрофия) кожи на участках с истощающей жировой, соединительной и мышечной ткани, что приводит к появлению необычно блестящих, гладких или затвердевших участков кожи, которые могут прилипать к подлежащим тканям. На пораженных участках могут возникать открытые язвы из-за снижения поступления насыщенной кислородом крови к тканям (ишемия). Язвы могут быть хроническими и медленно заживающими. Глубокие язвы вокруг ахиллова сухожилия и, реже, на локтях, очень характерны для синдрома Вернера.
Многие больные с синдромом Вернера также имеют дополнительные кожные аномалии. На коже рук и ног может наблюдаться гиперпигментация или гипопигментация и/или аномальное расширение определенных мелких нижележащих кровеносных сосудов, вызывая соответствующее покраснение (телеангиэктазии). Кроме того, кожа ладоней, подошв и над некоторыми выступающими суставами, такими как локти и колени, может стать необычно утолщенной (гиперкератоз) и иметь тенденцию к развитию язв из-за разрушения поверхностных тканей.
Из-за атрофических изменений кожи и подлежащих тканей в области лица больные могут иметь характерный «защемленный» вид лица, включая:
- необычно выступающие глаза;
- жесткие уши, утратившие эластичность;
- тонкий клювообразный и сплющенный нос.
Преждевременное поседение и выпадение волос придают характерный вид. Согласно сообщениям в медицинской литературе, у большинства пациентов с синдромом Вернера преждевременное старение проявляется примерно в возрасте от 30 до 40 лет.
Синдром Вернера также обычно характеризуется преждевременным началом старческой катаракты, состояния, при котором происходит потеря прозрачности линз глаз. У больных с синдромом Вернера катаракта обычно поражает оба глаза (двусторонняя) и внезапно возникает в течение третьего или четвертого десятилетия жизни. (Старческая катаракта обычно развивается у людей старше 50 лет). В некоторых случаях могут присутствовать и другие аномалии в глазах, такие как накопление отложений кальция в прозрачной области перед глазами (кальцификация роговицы), воспаление средних и внутренних слоев глаз (хориоретинит), дегенерация нервных клеток (палочек и колбочек) сетчатки, реагирующих на свет (пигментный ретинит), и/или прогрессирующая дегенерация центральной области сетчатки (старческая дегенерация желтого пятна/возрастная макулярная дегенерация). Степень связанного с этим ухудшения зрения зависит от степени тяжести и/или комбинации присутствующих аномалий глаз.
Приблизительно у 70 процентов заболевших на момент постановки диагноза развился инсулиннезависимый (или 2-го типа) сахарный диабет. Инсулиннезависимый сахарный диабет — это нарушение обмена веществ, характеризующееся резистентностью к действию гормона инсулина и аномальной секрецией инсулина поджелудочной железой, что приводит к повышению уровня простого сахара (глюкозы) в крови. (Инсулин регулирует уровень глюкозы в крови, способствуя перемещению глюкозы в клетки для производства энергии.) Эта форма диабета обычно развивается у здоровых людей в возрасте от 50 до 60 лет. Однако у больных с синдромом Вернера состояние может проявиться примерно к 35 годам. У больных может не быть явных симптомов на момент постановки диагноза или у них может наблюдаться учащенное мочеиспускание (полиурия), чрезмерная жажда (полидипсия), усиление голода (полифагия) и/или другие характерные симптомы. Кроме того, люди с этой формой диабета могут быть предрасположены к диабетической коме из-за резко сниженного уровня жидкости в клетках (гиперосмолярная диабетическая кома). Согласно сообщениям в медицинской литературе, хотя инсулинозависимый сахарный диабет может быть связан с определенными долгосрочными осложнениями, такими как повреждение нервов (диабетическая нейропатия), нарушение функции почек (диабетическая нефропатия) и повреждение кровеносных сосудов сетчатки (диабетическая ретинопатия), о таких осложнениях у пациентов с синдромом Вернера не сообщалось.
Синдром Вернера также характеризуется тяжелым, прогрессирующим, часто широко распространенным утолщением и потерей эластичности стенок артерий (артериосклероз). В некоторых пораженных артериях могут наблюдаться аномальные скопления отложений кальция в средней оболочке артерий и прогрессирующее разрушение и замещение мышечных и эластических волокон артерий фиброзной тканью (артериосклероз Менкеберга). Артерии, пораженные этой формой артериосклероза, могут включать те, которые транспортируют богатую кислородом кровь к сердечной мышце (коронарные артерии) или определенным артериям ног (заболевание периферических сосудов). Артериосклероз периферических кровеносных сосудов может вызвать или усугубить истощение кожи (атрофию) и изъязвления (язвы). Кроме того, аномальные отложения кальция могут накапливаться в определенных сердечных клапанах, например, клапане, расположенном там, где главная артерия тела (аорта) выходит из нижней левой камеры сердца (аортальный клапан), и клапане, расположенном между левой верхней и нижней камерами сердца (митральный клапан). Прогрессирующий артериосклероз может привести к:
- эпизодам боли в груди из-за недостаточного поступления кислорода в сердечную мышцу (приступы стенокардии);
- прогрессирующей неспособности сердца эффективно перекачивать кровь к легким и остальным частям тела (сердечная недостаточность);
- локализованной потере сердечной мышцы, вызванная нарушением ее кровоснабжения (инфаркт миокарда или сердечный приступ);
- и/или другим потенциально опасным для жизни осложнениям.
Больные с синдромом Вернера также имеют повышенную предрасположенность к раку. Наиболее распространенными новообразованиями при синдроме Вернера являются рак щитовидной железы, за которыми следуют рак пигмент-продуцирующих клеток кожи и слизистых оболочек (злокачественная меланома), рак защитных мембран, окружающих головной и спинной мозг (менингиома), опухоли, возникающие внутри мягкие ткани и кости (саркомы и остеосаркомы), саркомы мягких тканей, первичные опухоли костей и лейкоз/миелодисплазия.
Из-за прогрессирующего атеросклероза, злокачественных новообразований и/или других связанных нарушений многие пациенты с синдромом Вернера могут испытывать опасные для жизни осложнения примерно к четвертому или пятому десятилетию жизни.
Причины
Синдром Вернера передается по аутосомно-рецессивному признаку. Человеческие черты, в том числе классические генетические заболевания, являются продуктом взаимодействия двух генов, одного от отца, а другого от матери.
Рецессивные генетические расстройства возникают, когда человек наследует аномальный ген от каждого родителя. Если человек получает один нормальный ген и один патологический ген болезни, он будет носителем заболевания, но обычно бессимптомным. Риск для обоих родителей-носителей передать аномальный ген и, следовательно, иметь больного ребенка, составляет 25% при каждой беременности. Риск иметь ребенка-носителя, как и родителей, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей, составляет 25%. Риск одинаков для мужчин и женщин.
Родители некоторых людей с синдромом Вернера были близки по крови (кровные родственники). В этих случаях, если оба родителя несут один и тот же ген заболевания, существует более высокий, чем обычно, риск того, что их дети могут унаследовать два гена болезни, необходимые для развития заболевания.
Синдром Вернера вызывается аномальными (патологическими) изменениями (мутациями) в гене WRN. У людей с этим заболеванием выявлено более 80 различных мутаций гена WRN.
WRN ген кодирует белок «геликаза», что позволяет предположить, что нарушение метаболизма ДНК участвует в преждевременном старении, наблюдаемом у людей с этим заболеванием. Метаболизм относится к химическим процессам, происходящим в тканях тела. ДНК или дезоксирибонуклеиновая кислота несет генетический код в клетках, имеет спиральную лестничную структуру и состоит из цепей определенных химических групп. Считается, что белки «геликазы» ДНК способствуют «раскручиванию» ДНК во время определенных клеточных действий, таких как восстановление поврежденной ДНК и разделение идентичных хромосом (хромосомная сегрегация) на две «дочерние клетки» во время деления и размножения клеток. Исследователи предполагают, что синдром Вернера возникает из-за полной потери функции белка геликазы, кодируемого WRN ген. Специфическая функция белка геликазы в предотвращении преждевременного старения остается неясной.
Однако в ходе лабораторных (in vitro) исследований образцов клеток кожи (культивируемых фибробластов человека) исследователи продемонстрировали, что клетки людей без заболевания могут размножаться примерно в 60 раз («удвоение популяции»), тогда как фибробласты с синдромом Вернера могут воспроизводиться только в примерно в 20 раз. На основании таких результатов некоторые исследователи предположили, что WRN по сути является «счетным геном», регулирующим общее количество раз, которое клетки человека могут делиться и воспроизводиться. Такие исследователи предполагают, что мутации гена WRN могут привести к преждевременному ингибированию процессов репликации ДНК (синтеза) и раннему клеточному старению — событиям, которые обычно происходят позже в нормальных стареющих клетках человека.
Исследователи также наблюдали высокую частоту хромосомных аномалий (например, случайные транслокации) в культивируемых клетках кожи (фибробластах) и культивируемых лейкоцитах (лимфоцитах), полученных из определенных клеточных линий (клонов) у людей с синдромом Вернера. Такие данные (иногда называемые «мозаицизмом с разнообразными транслокациями») предполагают, что «поломка хромосом» может быть характерной чертой или играть определенную роль в процессе болезни. Однако конкретные последствия таких открытий остаются неизвестными, и требуются дальнейшие исследования.
Затронутые группы населения
Синдром Вернера — редкое заболевание, которое поражает в равной степени мужчин и женщин. Поскольку расстройство было впервые описано в медицинской литературе в 1904 году (О. Вернером), было зарегистрировано более 800 случаев. Частота расстройства оценивается от 1 до 20 на миллион человек. Хотя определенные связанные признаки присутствуют в детстве, в период полового созревания и в молодом возрасте, расстройство чаще всего выявляется в третьем или четвертом десятилетии жизни.
Близкие по симптомам расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Вернера. Сравнения могут быть полезны для дифференциальной диагностики:
- Прогерия (синдром Хатчинсона-Гилфорда или синдром преждевременного старения) — очень редкое детское заболевание, характеризующееся преждевременным старением, низким ростом и характерными чертами лица. Первичные симптомы расстройства связаны с процессом старения. Дети с синдромом Хатчинсона-Гилфорда стареют очень быстро и в молодом возрасте страдают расстройствами пожилого возраста. Примерно к 10 годам большинство детей с синдромом преждевременного старения достигают роста среднего 3-летнего ребенка. Часто возникает артрит, который поражает костные суставы в детском и подростковом возрасте.
- Синдром Готтрона (акрогерия) — легкая наследственная форма преждевременного старения (прогерия), характеризующаяся аномально маленькими руками и ногами с тонкой и нежной кожей. С младенчества дети с заболеванием кажутся старше своего фактического возраста. Кожа на руках и ногах необычайно тонкая и похожа на пергамент. Кисти и стопы остаются аномально маленькими и в зрелом возрасте.
- Синдром Де Барси — редкое заболевание, передающееся по аутосомно-рецессивному генетическому признаку. Основные характеристики включают дегенерацию эластической ткани кожи (эластолизис), непроизвольные движения рук и ног (атетоз), помутнение роговицы глаз, большие выступающие уши и потерю мышечного тонуса. Другие симптомы могут включать необычную гибкость мелких суставов, выступающий наружу лоб и/или низкий рост. Потеря эластичности кожи приводит к старению и морщинистости.
- Синдром Малвихилла-Смита — чрезвычайно редкое наследственное заболевание, связанное с преждевременным старением. Он характеризуется низким ростом, маленькой головой (микроцефалия), старением лица, потерей жировых слоев под кожей, множественными пигментными пятнами на коже (невусами), потерей слуха и/или нарушениями иммунной системы. Это заболевание может возникнуть в детском или подростковом возрасте. Синдром Малвихилла-Смита описан только у четырех человек в медицинской литературе.
- Синдром Шторма — чрезвычайно редкое наследственное заболевание, связанное с преждевременным старением и заболеваниями сердца. Другие симптомы в подростковом возрасте могут включать потерю бровей и ресниц, а также истончение и поседение волос на коже головы. Кожа на руках и лице становится стянутой, морщинистой. Некоторые больные также испытывают боль и нарушение функции суставов.
- Комбинированный дефект факторов роста (синдром Вернера из-за комбинированного дефицита факторов роста) — чрезвычайно редкое наследственное заболевание. Симптомы этого расстройства схожи с симптомами синдрома Вернера и включают потерю жировых слоев на руках и ногах, клювообразный нос, небольшую челюсть и/или узкий рот. Другие симптомы могут включать нарушение функции суставов, плоскостопие и тонкую стянутую кожу.
- Синдром Ротмунда-Томсона — наследственное заболевание кожи, характеризующееся аномальным покраснением кожи, вызванным закупоркой мелких кровеносных сосудов (капилляров). Большинство больных с синдромом Ротмунда-Томсона имеют маленький рост, потерю мышечной массы и красный цвет кожи. Некоторые больные аномально чувствительны к свету (светочувствительность) и могут развить катаракту в подростковом возрасте. Другие симптомы могут включать недоразвитые зубы и ногти, необычно маленькие руки и ноги, деформированные или отсутствующие большие пальцы рук и/или редкие и преждевременные седые волосы или облысение.
Диагностика
В некоторых случаях синдром Вернера можно распознать клинически уже примерно в возрасте 15 лет на основании тщательной клинической оценки, характерных физических данных (например, отсутствие скачка роста в период полового созревания, низкий рост, низкий вес) и внимательного отношения к истории пациента и семьи. Однако заболевание часто не может быть распознано или подтверждено до третьего или четвертого десятилетия жизни после того, как будут отмечены определенные характерные симптомы и признаки (например, преждевременное поседение и выпадение волос, характерный голос, потеря подкожной ткани, мышечная атрофия, изменения кожи, двусторонняя старческая катаракта и проч.).
Для обнаружения, подтверждения или характеристики определенных аномалий, потенциально связанных с заболеванием, могут проводиться специализированные визуализационные исследования и лабораторные тесты. Например, глазные специалисты (офтальмологи) могут регулярно контролировать больных на предмет развития катаракты с помощью определенных мер, таких как использование специального инструмента, который позволяет визуализировать внутреннюю часть глаз (офтальмоскоп). При обнаружении катаракты можно использовать микроскоп с подсветкой (щелевую лампу) для исследования внутренних структур передних отделов глаз, что позволяет офтальмологам определить конкретное расположение и степень катаракты.
Дополнительные исследования могут включать мониторинг уровня сахара в крови для обеспечения быстрого выявления сахарного диабета, сканирование костей и анализы крови на остеопороз и/или другие исследования. Кроме того, также могут проводиться тщательные кардиологические обследования и постоянный мониторинг (например, клинические осмотры, рентгеновские исследования, специализированные сердечные обследования) для оценки связанных сердечно-сосудистых аномалий и определения соответствующего лечения заболевания. Лица с синдромом Вернера также должны регулярно наблюдаться по мере необходимости, чтобы обеспечить быстрое обнаружение и соответствующее лечение определенных злокачественных новообразований или доброкачественных опухолей, которые могут возникать в связи с заболеванием (например, остеосаркома, менингиома).
В некоторых случаях специализированные лабораторные анализы могут проводиться на культивируемых клетках кожи (фибробластах) больных пациентов, демонстрируя аномально сниженную репликацию фибробластов синдрома Вернера. Оценка хромосомного состава (кариотипа) в ядрах культивируемых фибробластов и некоторых лейкоцитов (лимфоцитов) может выявить высокую частоту определенных хромосомных перестроек (мозаицизм неоднородной транслокации). Кроме того, по мнению нескольких исследователей, анализы мочи могут выявить повышенный уровень гиалуроновой кислоты, сложного углевода, который присутствует в промежутках между определенными клетками (межклеточные пространства) в определенных соединительных тканях. Последствия этого открытия не поняты.
Подтверждение клинического диагноза синдрома Вернера может быть достигнуто путем молекулярного исследования гена WRN. Молекулярное секвенирование гена WRN для выявления мутаций, вызывающих заболевание, а также биохимическое исследование для количественного определения количества белка WRN, производимого клетками, доступно на клинической основе.
Стандартные методы лечения
Лечение синдрома Вернера направлено на устранение конкретных симптомов, которые проявляются у каждого человека. Для лечения расстройства могут потребоваться скоординированные усилия группы специалистов, которым может потребоваться систематическое и всестороннее планирование лечения пострадавшего. К таким специалистам могут относиться терапевты; врачи, занимающиеся диагностикой и лечением заболеваний скелета, мышц, суставов и других родственных тканей (ортопеды); врачи, которые диагностируют и лечат патологии сердца и его основных кровеносных сосудов; окулисты (офтальмологи); врачи, занимающиеся диагностикой и лечением нарушений эндокринной системы (эндокринологи); и/или другие специалисты здравоохранения.
Специфические методы лечения пациентов с синдромом Вернера являются симптоматическими и поддерживающими. Согласно сообщениям в медицинской литературе, сахарный диабет, как правило, протекает в легкой форме, и его часто можно лечить с помощью диетических изменений и приема соответствующих лекарств внутрь для снижения повышенного уровня сахара (глюкозы) в крови (пероральные гипогликемические препараты).
У больных с катарактой лечение может включать хирургическое удаление помутневшего хрусталика и имплантацию заменяющего хрусталика (интраокулярной линзы) или назначение корректирующих очков или контактных линз. Некоторые врачи сообщают, что пациенты с синдромом Вернера могут иметь значительно повышенный риск отделения слоев хирургической раны (расхождение раны) и/или других осложнений (например, декомпенсации эндотелия роговицы). Поэтому эти врачи рекомендуют соблюдать особые меры предосторожности во время таких хирургических процедур (например, делать небольшие хирургические разрезы, избегать применения местного или системного кортизона).
У больных с синдромом Вернера меры по лечению артериосклероза и связанных с ним сердечно-сосудистых аномалий являются симптоматическими и поддерживающими. Например, у пациентов с приступами боли в груди из-за недостаточного поступления кислорода в сердечную мышцу (приступы стенокардии) лечение может включать использование определенных лекарств, которые могут помочь минимизировать или контролировать такие симптомы.
Если доброкачественные или злокачественные опухоли развиваются в связи с синдромом Вернера, соответствующие меры лечения могут варьироваться в зависимости от конкретного типа имеющейся опухоли; доброкачественная или злокачественная опухоль; тип, стадия и/или степень заболевания; и/или другие факторы. В зависимости от таких факторов методы лечения могут включать хирургическое вмешательство, использование определенных противоопухолевых препаратов (химиотерапию), лучевую терапию и/или другие меры.
Больным с синдромом Вернера и их семьям рекомендуется генетическое консультирование.
Прогноз
Прогноз неблагоприятный. Средняя продолжительность жизни пациентов с синдромом Вернера составляет 46 лет. Смерть обычно наступает в возрасте 30-50 лет из-за атеросклероза или злокачественных опухолей. Умелое медицинское введение пациента может увеличить продолжительность его жизни; Был описан один пациент, который дожил до возраста 68 лет и умер от острой сердечной недостаточности.