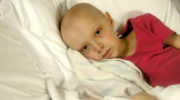
Что такое синдром Беквита-Видемана?
Синдром Беквита-Видемана (СБВ) — наиболее распространенное заболевание, связанное с чрезмерным ростом и предрасположенностью к раку. СБВ вызывается изменениями на хромосоме 11p15.5 и характеризуется широким спектром симптомов и физических признаков, которые варьируются по диапазону и степени тяжести от человека к человеку. Сопутствующие признаки включают массу тела при рождении выше среднего (большая для гестационного возраста), усиленный рост после рождения (макросомия), большой язык (макроглоссия), увеличение определенных внутренних органов (органомегалия) и дефекты брюшной стенки (омфалоцеле, пупочная грыжа или диастаз прямых мышц живота).
Синдром Беквита-Видемана также может быть связан с низким уровнем сахара в крови в первые несколько дней жизни (неонатальная гипогликемия) или позже, что приводит к стойкому низкому уровню сахара в крови (гиперинсулинизм), характерным бороздкам в мочках ушей, лицевым аномалиям, увеличению одной стороны или структуры тела, приводящее к неравномерному (асимметричному) росту и повышенному риску развития некоторых видов рака у детей, чаще всего опухоли Вильмса (опухоль почки) и гепатобластомы (опухоли печени).
Синдром Беквита-Видемана недавно был переклассифицирован как спектр Беквита-Видемана, поскольку клинические проявления могут варьироваться от пациента к пациенту. Примерно у 80% людей с СБВ изменения происходят случайным образом (спорадически). Семейная передача (наследственные формы) встречается примерно у 5-10% пациентов с СБВ. Около 14% пациентов с СБВ имеют неизвестную причину диагноза. СБВ поражает как минимум одного из 10 340 живорожденных. Исследователи определили, что синдром возникает в результате различных аномалий, влияющих на нормальную, правильную экспрессию определенных генов, которые контролируют рост в пределах определенной области хромосомы 11 (критическая область BWS).
Признаки и симптомы
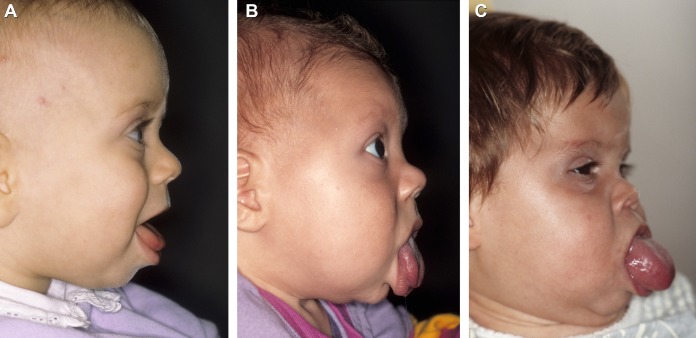
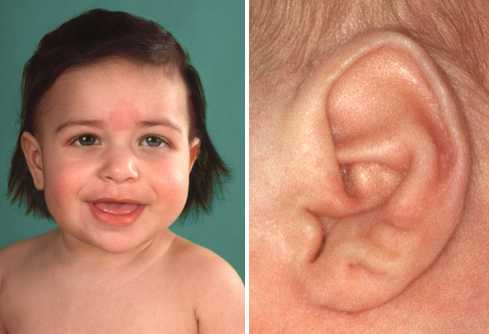
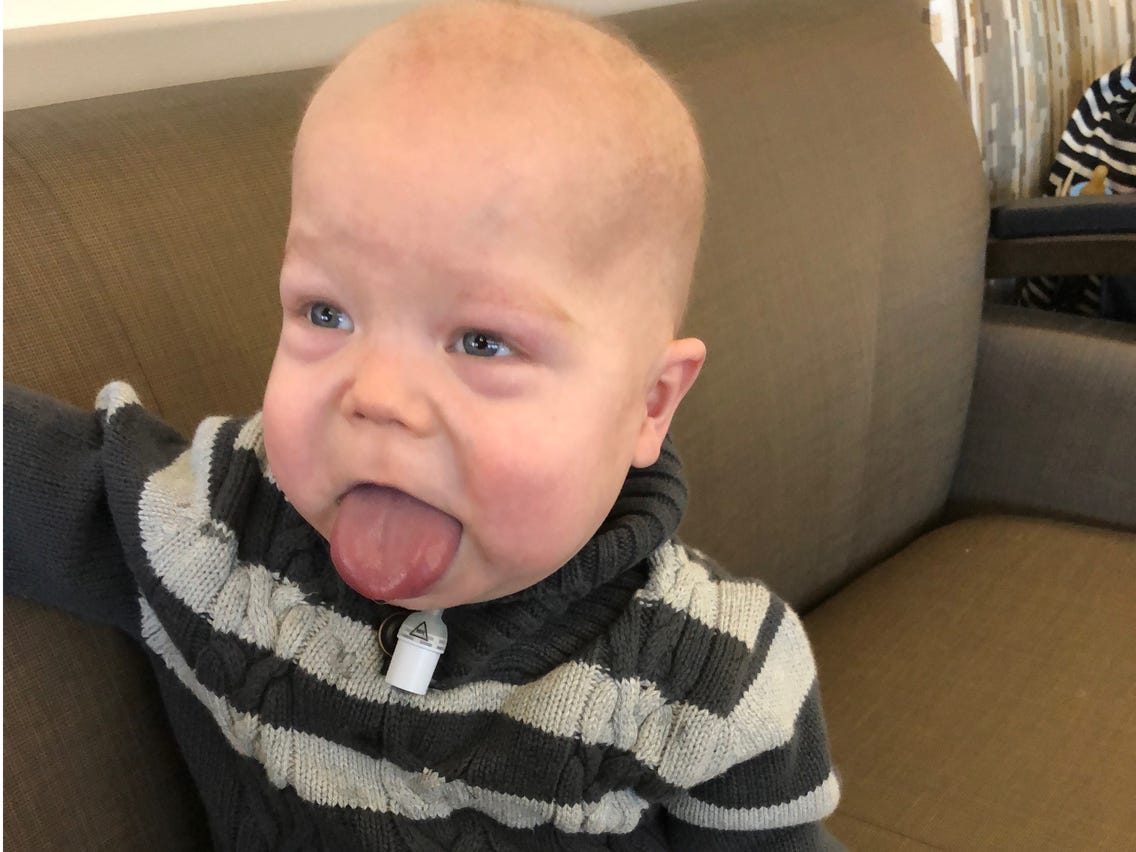
Фенотипические особенности синдрома сильно различаются от человека к человеку, что позволяет поставить клинический диагноз на основе результатов физического осмотра и молекулярной диагностики на основе генетического тестирования. Иногда клинические и молекулярные диагнозы не совпадают, потому что клинически пациенты могут не иметь многих заметных физических характеристик синдрома Беквита-Видемана, даже если у них есть изменения в критической области BWS на основе генетического тестирования. Некоторые особенности больных могут казаться легкими, в то время как другие — более значительными. У больных могут быть не все перечисленные симптомы. Диапазон клинических признаков, вызванных изменениями на хромосоме 11p15.5, был переопределен как спектр Беквита-Видемана.
К особенностям синдрома Беквита-Видемана относятся:
- большой вес и длина при рождении (макросомия);
- разрастание одной стороны или одной части тела (гемигипертрофия/гемигиперплазия);
- увеличенный язык (макроглоссия);
- низкий уровень сахара в крови (гипогликемия) в период новорожденности и иногда хроническая гипогликемия (из-за гиперинсулинизма);
- дефекты брюшной стенки (например, пупочная грыжа или омфалоцеле, когда кишечник, а иногда и другие органы брюшной полости выступают из брюшной стенки за пределы тела);
- увеличение органов брюшной полости, таких как почки, печень и поджелудочная железа;
- ямки или складки в мочке уха или за ухом;
- повышенный риск развития некоторых видов рака в детстве (большинство из которых можно вылечить с помощью надлежащего лечения).
Пациенты с СБВ могут иметь повышенный риск развития некоторых видов рака. Эмбриональный рак встречается примерно у 8% пациентов с синдромом Беквита-Видемана. Наиболее распространенными типами опухолей являются опухоль Вильмса (опухоль почки), гепатобластома (опухоль печени), нейробластома (опухоль нервных клеток), рабдомиосаркома (опухоль мягких тканей) и карцинома надпочечников. Общий риск опухоли наиболее высок в течение первых двух лет жизни.
Многие клинические признаки СБВ становятся менее очевидными с возрастом, и у многих взрослых наблюдается нормальный рост и внешний вид. Неврологическое (мозговое) развитие, по-видимому, не затрагивается при заболевании, если только оно не связано с длительной нелеченной неонатальной гипогликемией, крайней недоношенностью или хромосомной дупликацией. Взрослые пациенты могут иметь медицинские проблемы, связанные с этими клиническими особенностями, или требовать хирургического вмешательства в раннем детстве. Большинство особенностей у взрослых с СБВ, таких как проблемы с почками и боль в спине, являются следствием педиатрических проблем. Однако необходимы дополнительные исследования, чтобы определить взаимосвязь между особенностями взрослых с синдромом Беквита-Видемана и педиатрическими симптомами.
Причины и факторы риска
Этиология синдрома Беквита-Видемана сложна. Около 80-90% пациентов имеют известную молекулярную аберрацию, которая влияет на регуляцию группы импринтированных генов, участвующих в прогрессировании клеточного цикла и контроле соматического роста, расположенных в хромосоме 11p15.5. Геномный импринтинг — это регулируемый эпигенетикой процесс, посредством которого экспрессируется только одна копия гена в зависимости от пола родителя, несущего аллель (различные формы одного и того же гена). Две контрольные области импринтинга (IC1 и IC2) регулируют экспрессию гена из области 11p15. Обычно метилирование (подавление экспрессии гена) происходит в отцовском аллеле на IC1 и материнском аллеле на IC2. У человека с СБВ чаще всего описываются следующие молекулярные дефекты:
- Утрата метилирования по IC2 материнского аллеля в 50-60% случаев;
- Отцовская монородительская изодисомия 11p15 в 20-25% случаев;
- Увеличение метилирования по IC1 материнского аллеля в 5-10% случаев;
- Аутосомно-доминантные материнские точечные мутации в CDKN1C (регулируются IC2) в 5% спорадических случаев и 40% семейных случаев;
- Хромосомные перестройки (дупликации, транслокации, делеции или инверсии) менее чем в 1% случаев;
- Неизвестный молекулярный дефект в 10-15% случаев.
Затронутые популяции
СБВ поражает мужчин и женщин в равных количествах. По оценкам, расстройство встречается у 1 из 10340 человек в общей популяции. Поскольку у людей с легкой степенью поражения синдром может не диагностироваться, поэтому трудно определить истинную частоту заболеваемости среди населения в целом.
Не существует особого повышенного риска СБВ в популяциях определенной расы/этнической принадлежности, хотя клинические проявления могут варьироваться в зависимости от группы.
Исследования показывают, что пациенты, зачатые с помощью вспомогательных репродуктивных технологий (ВРТ), таких как экстракорпоральное оплодотворение (ЭКО) и/или интрацитоплазматическая инъекция сперматозоидов (ИКСИ), могут подвергаться большему риску развития нарушений в результате геномного импринтинга (например, BWS), чем население в целом. Недавнее исследование выявило десятикратное увеличение риска заболевания у пациентов, зачатых с помощью АРТ, с распространенностью у одного из 1126 пациентов. Большинство пациентов с синдромом Беквита-Видемана, зачатых с помощью АРТ, имеют болезнь из-за IC2. Необходимы дополнительные исследования, чтобы определить точную связь между такими технологиями и развитием синдрома.
Исследования также показали, что частота беременностей близнецами чаще встречается в популяции СБВ, чем в общей популяции. Тем не менее, близнецы с СБВ, как правило, имеют разную степень тяжести, что затрудняет для врачей диагностику и лечение близнецов с заболеванием.
Близкие расстройства
Симптомы следующих заболеваний могут быть аналогичны симптомам синдрома Беквита-Видемана. Сравнения могут быть полезны для дифференциальной диагностики:
- Синдром Симпсона-Голаби-Бемеля — Х-сцепленное рецессивное генетическое заболевание, вызванное мутациями в генах GPC3 или GPC4. Синдром Симпсона-Голаби-Бемеля характеризуется чрезмерным ростом до и после рождения (макросомия); особым внешним видом лица, включая широко расставленные глаза (гипертелоризм), грубые черты лица, аномально большой рот (макростомия), большую голову (макроцефалия), аномально большой язык (макроглоссия) и умственную отсталость от легкой до тяжелой.
- Синдром Перлмана — чрезвычайно редкое генетическое заболевание из-за рецессивных мутаций в гене DIS3L2, расположенном на хромосоме 2q37.1. Синдром Перлмана характеризуется чрезмерным ростом до и после рождения (макросомия), отличительными чертами лица, аномально увеличенными внутренними органами (органегалия), наличием фрагментов (остатков) эмбриональной ткани в почке (нефробластоматоз) и предрасположенностью к развитию опухоли Вильмса.
- Синдром Сотоса — редкое генетическое заболевание из-за спорадических мутаций гена NSD1, расположенного на хромосоме 5q35.3. Синдром Сотоса является аутосомно-доминантным заболеванием, что означает, что для поражения пациента необходима только одна копия мутировавшего гена. Синдром Сотоса характеризуется чрезмерным ростом как до, так и после рождения (внутриутробно и в послеродовой период). Новорожденные обычно демонстрируют развитый рост костей, аномально большие руки и/или ступни и характерные черты лица. Характерные лицевые аномалии могут включать необычно большую голову (макроцефалия), которая может казаться удлиненной (долихоцефалия), с аномально выступающим лбом (выступающий лоб); широко расставленные глаза (глазной гипертелоризм); наклонные вниз складки век (глазные щели), сильно изогнутое небо рта, выпячивание нижней челюсти (прогнатизм); и/или заостренный подбородок. У пораженных младенцев и пациентов могут также наблюдаться нарушения развития, включая задержки в достижении этапов развития (например, сидение, ползание и ходьба), задержки в координации мышечной и умственной деятельности (психомоторная отсталость) и задержки в языковых навыках.
- Синдром Уивера, также известный как синдром Уивера-Смита, является чрезвычайно редким аутосомно-доминантным заболеванием, вызванным мутациями в гене EZH2, расположенном на хромосоме 7q36.1. Синдром Уивера характеризуется ускоренным ростом. Черты лица пораженного пациента могут включать высокий широкий лоб, обычно круглое лицо, широко расставленные глаза (глазной гипертелоризм) и аномально маленькую челюсть. У пациентов часто наблюдается повышенный мышечный тонус (гипертонус) и проблемы с суставами.
Диагностика
Пациентам с синдромом Беквита-Видемана может быть поставлен диагноз как до, так и после рождения с помощью физического обследования (клинический диагноз) и/или генетического тестирования (молекулярный диагноз).
В некоторых случаях определенные процедуры могут быть выполнены до рождения (пренатально) для выявления расстрйоства. Например, ультразвуковая визуализация может позволить оценить размер органа и общий размер развивающегося плода и потенциально выявить другие признаки, которые могут указывать на спектр Беквита-Видемана. Особенности, которые могут быть обнаружены с помощью пренатальной визуализации, включают увеличение количества околоплодных вод вокруг плода (многоводие), увеличенную плаценту (плацентамегалия), омфалоцеле, увеличенную окружность живота, нефромегалию, макроглоссию и/или другие нарушения и аномалия. Наиболее частым пренатально обнаруживаемым признаком, который приводит к более высокому клиническому подозрению на СБВ, является омфалоцеле. Если есть подозрение на СБВ, доступно пренатальное обследование.
СБВ может быть диагностирован или подтвержден вскоре после рождения на основе тщательной клинической оценки, обнаружения характерных физических признаков (например, увеличение веса и длины, макроглоссия, дефекты брюшной стенки) и генетического тестирования критической области BWS.
Стандартные методы лечения
Лечение синдрома Беквита-Видемана направлено на устранение конкретных симптомов, которые проявляются у каждого человека. Лечение может потребовать согласованных усилий команды специалистов. Генетикам, педиатрам, пластическим хирургам, эндокринологам, нефрологам (специалистам по почкам), ортодонтам (стоматологам), пульмонологам, логопедам, детским онкологам и другим медицинским работникам может потребоваться систематическое и всестороннее планирование лечения пострадавшего ребенка.
У новорожденных с синдромом следует проводить регулярный мониторинг уровня глюкозы в крови, чтобы обеспечить быстрое обнаружение и лечение гипогликемии. Хотя неонатальная гипогликемия обычно бывает легкой и временной, ее раннее выявление и лечение имеют важное значение для предотвращения сопутствующих неврологических осложнений. Меры лечения могут включать введение глюкозы внутривенно, частые кормления, определенные лекарства (например, диазоксид или октреотид) и/или в некоторых случаях хирургическое вмешательство.
У многих младенцев с пупочной грыжей дефект может самопроизвольно исчезнуть примерно к одному году. Хирургическое вмешательство обычно не требуется, если только пупочная грыжа не становится все больше, не разрешается спонтанно (например, примерно к трем или четырем годам) и/или не связана с определенными осложнениями. Однако у новорожденных с омфалоцеле хирургическое лечение дефекта обычно требуется вскоре после рождения.
Подобно другим особенностям, связанным с синдромом Беквита-Видемана, макроглоссия может различаться по степени тяжести. Пациенты с макроглоссией подвержены повышенному риску обструктивного апноэ во сне, затруднений с кормлением, проблем с речью и потенциальных проблем с развитием челюсти. Пациентам с макроглоссией требуется поддержка многопрофильной бригады. Им следует пройти обследование кормления и сна, а также при необходимости проконсультироваться с пластическими хирургами и пульмонологами. Проблемы с кормлением, вызванные макроглоссией, могут потребовать поддержки специалистов по кормлению или диетологов. Лечение может включать использование специализированных сосков или временное введение назогастрального зонда. Речевые трудности могут потребовать поддержки логопеда. Пульмонолог может оценить степень влияния макроглоссии на дыхание и сон пациента. Полисомнография (исследование сна) может использоваться для оценки обструктивного апноэ во сне, обструкции дыхательных путей, сопротивления дыхательных путей, сильной десатурации, нарушения дыхания во сне и храпа. Постоянное положительное давление в дыхательных путях — это метод, используемый для поддержки детей с обструктивным апноэ во сне. Некоторым пациентам может быть сделана операция по уменьшению языка с целью улучшения дыхания, кормления и улучшения челюстей или пороков развития зубов из-за макроглоссии.
Пациентам с латерализованным разрастанием рекомендуется регулярное ортопедическое обследование. Некоторым пациентам со значительным латерализованным разрастанием конечностей может потребоваться хирургическая коррекция.
Кроме того, пациенты с синдромом Беквита-Видемана должны проходить регулярное ультразвуковое исследование брюшной полости и почек и измерение уровня альфа-фетопротеина в сыворотке в соответствии с рекомендациями, что позволяет раннее выявить и лечить определенные злокачественные новообразования, которые могут возникать в связи с синдромом Беквита-Видемана.
Если в связи с СБВ развивается опухоль, соответствующие меры лечения варьируются в зависимости от конкретной присутствующей опухоли, стадии и/или степени заболевания и/или других факторов. Методы лечения могут включать хирургическое вмешательство (например, нефронсохраняющую резекцию почки в случае опухоли Вильмса), использование определенных противоопухолевых препаратов (химиотерапию), лучевую терапию и/или другие меры.
Пациентам с сердечными, желудочно-кишечными и почечными нарушениями могут потребоваться определенные лекарства, хирургическое вмешательство или другое медицинское вмешательство. Этих пациентов следует направлять к соответствующим специалистам. Генетическое консультирование может быть полезным для пострадавших и их семей. Другое лечение симптоматическое и поддерживающее.
Прогноз
Прогноз варьируется в зависимости от тяжести клинической картины, молекулярного подтипа и своевременной диагностики состояния. В целом, у большинства пациентов с синдромом Беквита-Видемана будет нормальная продолжительность жизни. Взрослые с заболеванием будут иметь признаки, связанные с их педиатрическим фенотипом, с хорошим прогнозом, обеспечивающим раннее распознавание болезни, надлежащее упреждающее руководство и лечение осложнений, если они будут развиваться.
Осложнения
Ниже приведены некоторые из осложнений, наблюдаемых у пациентов:
- Перинатальные осложнения: гестационная гипертензия, преэклампсия, гестационный сахарный диабет, вагинальное кровотечение, многоводие, недоношенность и осложнения, связанные с макросомией (кефалогематома, травма плечевого сплетения или другие родовые травмы).
- Неонатальные осложнения: повышенный риск смерти в основном в результате осложнений, связанных с недоношенностью, макроглоссией (затрудненное дыхание и кормление), гипогликемией и, реже, кардиомиопатией.
- Другие осложнения: проблемы с речью, связанные с макроглоссией, когнитивные нарушения, связанные с конкретными хромосомными аномалиями и/или перинатальными осложнениями, высокий риск развития эмбриональных опухолей (особенно опухоли Вильмса и гепатобластомы), нефрокальциноз, нефролитиаз, кисты почек, рецидивирующие инфекции мочевыводящих путей, сколиоз, связанный с несоответствие длины ног, проблемы фертильности из-за крипторхизма и психосоциальные проблемы, связанные со страхом за состояние и осложнения.